Focus On AI Special ㅣ PRACTICAL
AI 혁신의 새 지평: 알파폴드(AlphaFold)와
알파미스센스(AlphaMissense)
Focus On I
Focus On III
분당서울대병원
서론
2024년 노벨 과학상은 인공지능(AI)이 과학 발전에 미친 심대한 영향을 조명하며, AI 기술의 중요성을 다시금 강조했다. 노벨 물리학상은 인공신경망을 활용한 기계학습 기초를 확립한 존 홉필드(John Hopfield)와 제프리 힌턴(Geoffrey Hinton)에게 수여되었으며, 노벨 화학상은 AI기반 단백질 구조 예측 모델 알파폴드(AlphaFold)를 개발한 데미스 허사비스(Demis Hassabis)와 존 점퍼(JohnJumper), 단백질 설계를 위한 계산 방법론을 제시한 데이비드 베이커(David Baker)에게 돌아갔다.
이 수상은 AI 기술이 생명과학과 의학 분야에서 새로운 지평을 열어가고 있음을 보여준다. 특히, 알파폴드와 같은 AI기반 단백질 구조 예측 기술은 단백질의 3차원 구조를 정확히 분석하며 유전자 변이가 단백질 기능에 미치는 영향을 파악해 진단과 치료를 지원하는 데 중요한 역할을 하고 있다. 이 글은 알파폴드 기술과 이를 확장한 알파미스센스가 생명과학 연구와 정밀 의학에서 어떻게 변화를 이끌고 있는지 살펴보고자 한다.
단백질 구조 예측의 발전 단백질은 아미노산 서열로부터 고유한 3차원 구조로 접히며, 이 구조가 단백질의 기능을 결정한다. 전통적으로 단백질 구조 예측은 X선 결정학, 핵자기공명(NMR) 분석,Cryo-EM 등 실험적 접근법에 의존해 왔다. 하지만 이러한 방법은 비용과 시간이 많이 소요되며, 막단백질이나 고분자 단백질과 같은 특수 단백질에는 적용이 어려운 한계가 있다.
이러한 한계를 극복하기 위해 등장한 알파폴드(Alpha-Fold)는 단백질 구조 예측에 혁신을 가져왔다. 알파폴드는 아미노산 서열만으로 단백질의 3차원 구조를 예측할 수있는 딥러닝 기반 모델로, 2020년 CASP14(Critical Assessment of protein Structure Prediction) 대회에서 기존 기술을 압도하는 성능을 기록했다. 알파폴드는 전통적인 생물물리학적 접근법에서 벗어나 예측 정확도를 획기적으로 향상시켰으며, 이를 통해 신약 개발과 단백질 연구의 속도를 대폭 단축했다.
트랜스포머(Transformer) 기술:알파폴드의 핵심 알파폴드의 기술적 기반은 2017년 구글이 발표한 논문 “Attention is All You Need”에서 제안된 트랜스포머(Transformer) 아키텍처이다. 트랜스포머는 딥러닝의 혁신적인 시퀀스-투-시퀀스(Sequence-to-Sequence) 모델로, 셀프 어텐션(Self-Attention) 메커니즘을 통해 시퀀스 내 요소 간의 관계를 정교하게 학습한다. 트랜스포머는 방대한 데이터를 병렬로 처리할 수 있어 자연어 처리(Natural Language Processing, NLP)뿐만 아니라 생명과학 분야에도 널리 활용되고 있다. 알파폴드2는 이 트랜스포머 아키텍처를 활용해 단백질 서열의 상호작용을 모델링하고, 이를 기반으로 고해상도 단백질 3차원 구조를 예측한다. 이러한 기술은 단백질 서열 데이터에서 숨겨진 생물학적 정보를 효과적으로 학습하며, 단백질 간 상호작용, 구조적 안정성, 그리고 약물 결합 가능성을 탐구하는데 큰 기여를 하고 있다.
알파폴드 알파폴드1은 딥러닝을 단백질 구조 예측에 도입하며 혁신의 시작을 알렸지만, 예측 정확도가 제한적이었고 데이터 요구량이 높아 실질적 응용에는 한계가 있었다. 그러나 알파폴드2는 트랜스포머 기술을 결합해 원자 수준에서 단백질 구조를 예측하는 데 성공하며 전환점을 마련했다. 이를통해 단백질 구조 데이터베이스를 확장하고, 신약 개발과 분자 진단 기술 발전을 가속화했다.
알파폴드3는 이러한 기술을 한 단계 더 발전시켜 복합 단백질 구조와 단백질-리간드, 단백질-DNA 등의 상호작용 예측까지 가능하게 했다. 이 시스템은 알파폴드2에서 사용된 Evoformer를 더 간단한 Pairformer 모듈로 교체하여 MSA (Multipe-sequence alignment) 처리량을 줄였고, diffusion 모듈을 사용하여 raw 원자 좌표를 직접 예측하는 방법을 사용하였다. 이 기술은 질병의 분자적 기전을 해독하거나, 특정 질병 마커와 약물의 결합 가능성을 분석하는 데 중요한 역할을 하고 있다. (그림 1)
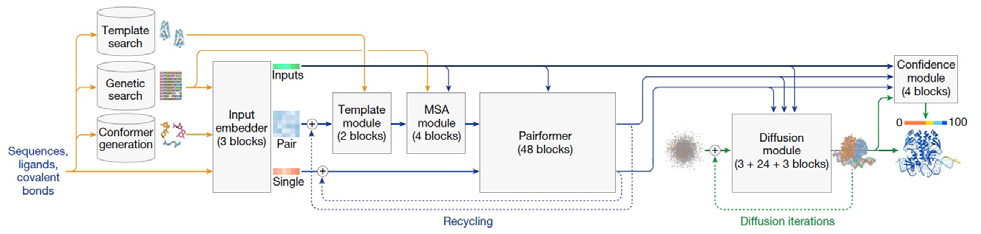 그림1
그림1
알파폴드3 구조 [figure from Abramson et al. Nature, https://doi.org/10.1038/s41586-024-07487-w]
과오 돌연변이(Missense Mutation)는 단백질 기능 이상을 유발해 질병의 원인이 되는 주요 변이이다. 그러나 현재 관찰된 400만 개 이상의 미스센스 변이 중 약 2%만이 병원성 또는 양성으로 분류되었으며, 나머지 98%는 임상적 중요성이 불분명한 상태로 남아 있다. 알파미스센스(AlphaMissense)는 이러한 문제를 해결하기 위해 알파폴드2의 단백질 구조 예측 기술을 확장하여 개발되었다. 알파미스센스는 단백질 서열과 구조적 정보를 통합적으로 분석하여, 단일 변이가 단백질 안정성, 상호작용, 또는 기능적 도메인에 미치는 영향을 정량적으로 평가한다. 이를 통해 병원성 여부를 판별하는 유해성 점수(Harmfulness Score)와 예측 결과의 신뢰도를 정량화한 신뢰도 점수(Confidence Score)를 제공한다. 알파미스센스는 최근 ClinVar 데이터베이스의 약 7100만개의 missense 변이를 분석해, 32%가 병원성일 가능성이 높고, 57%는 양성일 가능성이 높다고 평가했다. 이는 정밀의학과 유전 질환 연구에서 알파미스센스의 강력한 활용 가능성을 입증하는 사례이다. (그림 2)
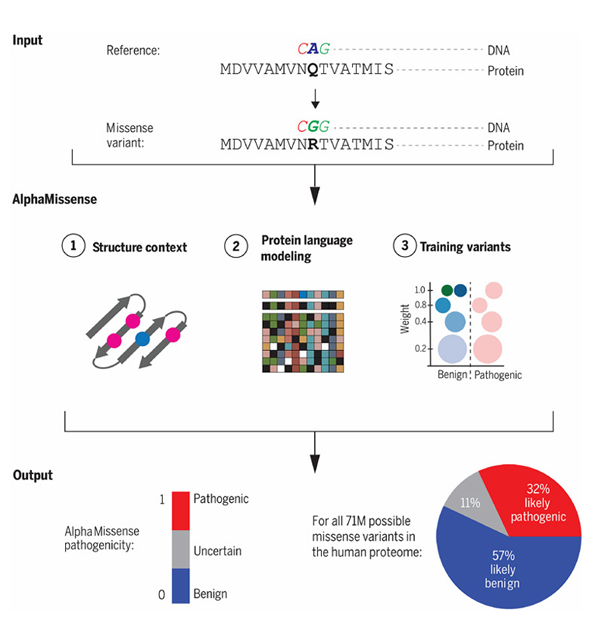 그림2
그림2
알파미스센스 개요 [figure from Cheng et al. Science, https://doi.org/10.1126/science.adg7492]
2024년 현재, 알파폴드 단백질 구조 데이터베이스는 2억개 이상의 예측 구조를 포함하며, 단백질 연구의 데이터 접근성을 획기적으로 확장했다. 앞으로 알파폴드와 알파미스센스는 기술의 정교화와 함께 예측 속도와 정확도를 더욱 높여갈 것이다. 특히, 단백질 상호작용 네트워크와 변이해석의 정밀도를 높이며, 신약 개발, 질병 진단, 정밀 의학 실현에 기여하는 핵심 도구로 자리 잡을 것이다. 아직 기능이 알려지지 않은 VUS(Variants of Uncertain Significance)변이들도 이 기술을 통해 기능이 밝혀지고, 질병의 분자적 기전 이해와 치료 전략 수립에 기여할 날이 머지않아 보인다.
[References]
1. Abramson, J., Adler, J., Dunger, J., Evans, R., Green, T., Pritzel, A., Ronneberger, O., Willmore, L., Ballard, A.
J., Bambrick, J., Bodenstein, S. W., Evans, D. A., Hung, C. C., O’Neill, M., Reiman, D., Tunyasuvunakool, K., Wu,
Z., Žemgulytė, A., Arvaniti, E., … Jumper, J. M. (2024). Accurate structure prediction of biomolecular interactions
with AlphaFold 3. Nature, 630(8016), 493–500. https://doi.org/10.1038/s41586-024-07487-w
2. Cheng, J., Novati, G., Pan, J., Bycroft, C., Žemgulyte, A., Applebaum, T., Pritzel, A., Wong, L. H., Zielinski,
M., Sargeant, T., Schneider, R. G., Senior, A. W., Jumper, J., Hassabis, D., Kohli, P., & Avsec, Ž. (2023). Accurate
proteome-wide missense variant effect prediction with AlphaMissense. Science, 381(6664). https://doi.
org/10.1126/science.adg7492
3. Jumper, J., Evans, R., Pritzel, A., Green, T., Figurnov, M., Ronneberger, O., Tunyasuvunakool, K., Bates, R.,
Žídek, A., Potapenko, A., Bridgland, A., Meyer, C., Kohl, S. A. A., Ballard, A. J., Cowie, A., Romera-Paredes, B., Nikolov,
S., Jain, R., Adler, J., … Hassabis, D. (2021). Highly accurate protein structure prediction with AlphaFold.
Nature, 596(7873), 583–589. https://doi.org/10.1038/s41586-021-03819-2
4. Senior, A. W., Evans, R., Jumper, J., Kirkpatrick, J., Sifre, L., Green, T., Qin, C., Žídek, A., Nelson, A. W. R., Bridgland,
A., Penedones, H., Petersen, S., Simonyan, K., Crossan, S., Kohli, P., Jones, D. T., Silver, D., Kavukcuoglu,
K., & Hassabis, D. (2020). Improved protein structure prediction using potentials from deep learning. Nature,
577(7792), 706–710. https://doi.org/10.1038/s41586-019-1923-7
5. Vaswani, A., Brain, G., Shazeer, N., Parmar, N., Uszkoreit, J., Jones, L., Gomez, A. N., Kaiser, Ł., & Polosukhin,
I. (2017). Attention Is All You Need. 31st Conference on Neural Information Processing Systems (NIPS 2017)
6. 남궁석, 알파폴드: AI 신약개발 혁신, BIOSPECTATOR, 2024
