씨젠의료재단 유전체연구소
NGS (차세대염기서열분석) 기술은 방대한 양의 데이터를 생성하며, 유전자 염기 서열, 복제 수, 발현량, 메틸화 (methylation) 등 다양한 요소를 측정할 수 있게 발전해왔다. 분석의 정확도를 높이기 위해 NGS에는 여러 AI 기술이 적극적으로 사용되고 있다. 이 글에서는 머신 러닝과 딥 러닝을 포함한 AI 기술이 유전체 분석에 어떻게 활용되고 있고, 향후 유망한 활용 분야는 어디일지 살펴본다.
NGS를 이용한 변이 검출은 기본적으로 어느 정도의 에러 가능성을 가지고 있다. 머신 러닝은 변이 호출 에러를 줄여 변이 호출 성능을 개선하는데 중요한 역할을 해왔다. 변이 호출 (variant calling)은 레퍼런스 서열과 다른 염기 정보가 있는지 확인하고, 노이즈 여부를 결정하는 과정을 말한다. 변이 검출 파이프라인 가이드라인 중 하나인 GATK best practice에서는 생식세포 변이 검출 분야에서 VQSR (Variant Quality Score Recalibration)이라는 가우시안 혼합 모델 (Gaussian mixture model)을 사용할 것을 권장하고 있다. VQSR은 GATK haplotypecaller를 통해 얻어진 검사실 샘플 변이 데이터와 인구집단 데이터에서 True call일 가능성이 높은 변이 정보 세트(HapMap3, 1000G Omni 등)를 기반으로 진실/거짓 세트를 만들고 이를 기반으로 학습시켜 변이 필터링 기준을 만든다. 그러나 최소 50개 이상의 WES 혹은 1개 이상의 WGS 데이터로 학습시키길 권장하고 있어, 기존에 생산한 데이터가 충분하지 않다면 하드필터링 방식 등 다른 방법을 고려하는 것이 좋다.
또한, GATK Best Practice에서 권장하는 다른 변이 검출 방식 중 하나인 CNNScoreVariant는 합성곱 신경망(CNN)을 기반으로 한다. 구글의 DeepVariant 역시 CNN을 기반으로 하며, 2020 FDA challenge에서 모든 종류의 변이 호출 정확성 면에서 SOTA (State of the Art)를 달성하여, 현재까지 가장 정확한 생식세포 변이 호출 알고리즘 중 하나에 속한다.
 그림1
그림1
Google Deep variant의 변이에 대한 입력 채널 정보. Deep variant는 mpileup된 이미지를 기반으로, 시퀀스, 품질, 방향, 변이 정보를 이미지화 시킨 후 이를 6개의 채널로 프로세싱하는 CNN 구조를 가진다.
Inception V3와 같은 CNN 기반 deep learning model을 이용해 변이 호출을 수행한다.
RNA sequencing 어플리케이션 중 차등 발현 유전자(Differential expression gene, DEG) 분석이나 Single Cell RNA sequencing과 같은 경우 유전자 발현량에 관련된 질환과의 패턴을 찾는 작업이 필요하다. 이 때 데이터의 차원 수는 분석하고자 하는 RNA transcript의 종류에 비례하여 증가한다. 모든 유전자를 타겟으로 할 경우 사람이 직관적으로 처리하기 어려운 방대한 양의 고차원 데이터가 생성된다. 이러한 복잡성 때문에 발현량과 관련된 패턴 분석은 머신 러닝과 딥 러닝 기술에 대한 의존도가 다른 분석에 비해 높은 편이다.
RNA sequencing 데이터는 주로 RNA의 발현량을 기반으로 유전자 간 상호 작용을 분석하거나, 유사한 발현 패턴을 가진 유전자 그룹을 탐색하는 데 주로 사용된다. 이때 고차원 데이터의 분석을 위해 차원 축소 기법인 PCA(Principal Component Analysis), t-SNE(t-Distributed Stochastic Neighbor Embedding), UMAP(Uniform Manifold Approximation and Projection)을 사용하여 데이터를 시각화 가능한 저차원으로 변환 가능하다. 예를 들어 Single cell RNA를 이용해 샘플 내 세포의 유형을 클러스터링을 하는 작업에는 기본적으로 UMAP, t-SNE, K-Means 등의 비지도 학습 (unsupervised learning) 기법이 널리 이용된다. 최근에는 Graph Neural Networks (GNN) 등을 이용해 기존에 알려진 유전자 발현량 정보를 결합하려는 시도도 이루어지고 있다. 또한, 공간 유전체학(Spatial genetics) 기술의 발전으로 현미경 이미지와 RNA 발현량 정보를 결합한 신경망 네트워크 분석도 활발히 진행되고 있다.
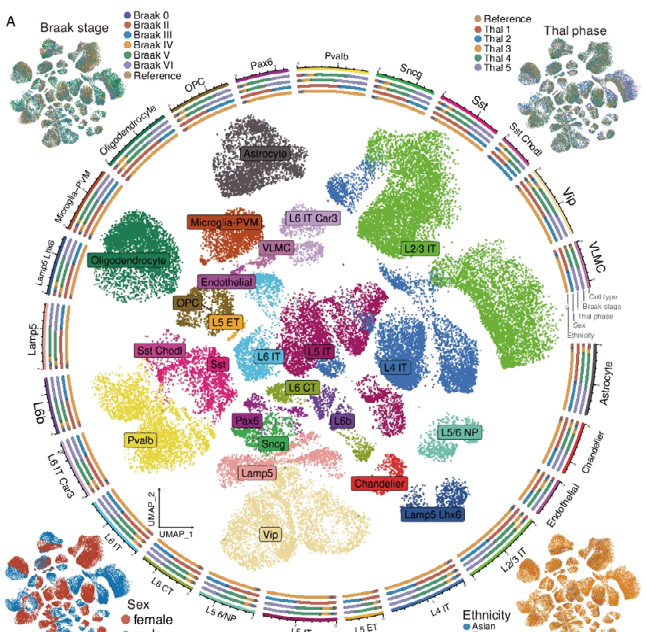 그림2
그림2
알쯔하이머 환자의 RNA 발현량 분석을 통한 UMAP 분석 결과
3. Large Language Model의 유전체 분석 활용 최근 가장 주목받고 있는 기술인 대형 언어 모델(Large Language Model, LLM)의 유전체 분석 분야 도입이 가시화되고 있다. LLM 기술은 크게 두 가지 방향으로 유전체 분석에 활용될 수 있다. 첫째, 인간 언어 해석 모델로서의 활용이다. 대형 언어 모델은 텍스트 기반으로 작성된 병원 전자의무기록 (Electronic Medical Record, EMR) 데이터를 기반으로 환자 정보를 레이블링하거나, 의학 문헌과 데이터베이스의 유전체 정보를 해석하는 데 활용될 수 있다. 예를 들어, OpenAI의 GPT-4, LLaMA, Mistral 등 다양한 모델들이 방대한 양의 정보를 학습한 상태로, 이러한 모델들을 활용하면 보다 정확한 유전 질환 진단이 가능해진다. 특히, 검색 증강 생성(Retrieval-Augmented Generation, RAG) 기법을 활용하면, 환자의 표현형과 유전 질환 간의 관계를 더욱 정확하게 분석할 수 있다. API (Application Programming Interface)나 오픈 소스 모델을 직접 도입하는 등 관련된 소스 코드 생태계가 빠른 속도로 정립되고 있어, 유전체 임상 검사실에서 대형 언어 모델의 도입은 얼마 남지 않은 상태로 보인다.
둘째, 염기서열 정보를 해석할 수 있는 모델로서의 활용이다. 염기서열 정보 해석은 아직 많은 연구가 필요하지만, DNA-BERT와 같은 모델은 DNA 염기서열을 처리하기 위한 대형 언어 모델의 가능성을 잘 보여주고 있다. DNABERT는 유전자 예측, 전사인자 결합 부위 예측, 스플라이싱 부위 인식, 프로모터 영역 감지 등에서 기존 모델들보다 뛰어난 성능을 보였다. 해당 연구 그룹은 DNA-BERT2, DNA-BERTS 등 후속 기반(foundation) 모델을 지속적으로 개발하고 있어, 앞으로 더 다양한 적용이 가능할 것으로 생각된다.
AI 기술은 NGS 분석과 관련된 여러 문제를 해결하고, 새로운 가능성을 열어줄 것이다. 그러나 AI 기술의 한계와 위험성도 존재한다. AI는 학습 데이터의 특성과 양에 따라 성능이 달라질 수 있으며, 종종 인간이 이해하기 어려운 오류를 발생시키기도 한다. 이러한 점을 고려하여 AI 기술을 임상 검사실에 도입할 경우 철저한 검증을 선행하고, 기존 분석 기술과 교차 검증을 시행할 것을 권장한다.
[References]
1. GATK VQSR: https://gatk.broadinstitute.org/hc/en-us/articles/360035531612-Variant-Quality-Score-Recalibration-VQSR
2. Poplin R, Chang PC, Alexander D, Schwartz S, Colthurst T, Ku A, Newburger D, Dijamco J, Nguyen N, Afshar PT, Gross SS, Dorfman L, McLean CY, DePristo MA. A universal SNP and small-indel variant caller using deep neural networks. Nat Biotechnol. 2018 Nov;36(10):983-987. doi: 10.1038/nbt.4235. Epub 2018 Sep 24. PMID: 30247488.
3. Wang J, Ma A, Chang Y, Gong J, Jiang Y, Qi R, Wang C, Fu H, Ma Q, Xu D. scGNN is a novel graph neural network framework for single-cell RNA-Seq analyses. Nat Commun. 2021 Mar 25;12(1):1882. doi: 10.1038/s41467-021-22197-x. Erratum in: Nat Commun. 2022 May 4;13(1):2554. doi: 10.1038/s41467-022-30331-6. PMID: 33767197; PMCID: PMC7994447.
4. Zhou Z, Wu W, Ho H, Wang J, Shi L, Davuluri RV, Wang Z, Liu H. DNABERT-S: Pioneering Species Differentiation with Species-Aware DNA Embeddings. ArXiv. 2024 Oct 22:arXiv:2402.08777v3. PMID: 38410647; PMCID: PMC10896361.
