이원의료재단
얼마전 블로그 하나를 보았는데, chatGPT가 열린지 이제 2년이 되었다는 뉴스였다. 생각해보면 그동안 AI 분야를 중심으로 엄청난 변화들이 있었던 것 같은데, 그를 따라잡기는 했는지 미궁 속에 있는 듯한 연말이다. 당시 chatGPT 개발사인 OpenAI 직원들은 최대 10만명 정도 사용할 거라고 예측했다던데, 어느새 유효 활성 이용자가 2억명을 넘어가고 있는걸 보니, 그 필드 전문가도 앞날을 예측하긴 어려운가보다.
이번 notable research에서는 올해 노벨화학상을 수상한 Demis Hassabis (CEO), John Jumper (수석연구원)이 포함된 구글 딥마인드 팀의 사이언스 지 논문을 가져왔다. 이 팀은 단백질 구조를 예측하는 구글 AlphaFold2를 개발한 공로로 수상하였는데, 이전에 X선 결정학, 극저온 현미경, NMR 등을 사용하여 실험적으로 분석하던 것을 딥러닝 알고리즘을 통해 혁신적으로 시간과 비용을 절감한 바를 인정받았다 (2021년 Nature 논문의 1저자, 교신저자).
아래는 메릴랜드대 세포생물학 및 분자유전학과 교수인 존 몰트의 주도로 2년마다 열리는 단백질 구조 예측 대회인 CASP13, CASP14에서 각각 선보인 AlphaFold, AlphaFold2의 성능에 대한 그래프이다. Global distance test 상 90점이면 실험적으로 밝힌 구조와 유사하다고 인정하는 바, AlphaFold2는 이전 십수년 간 도달하지 못했던 비약적인 성과를 이뤄냈다고 볼 수 있다.
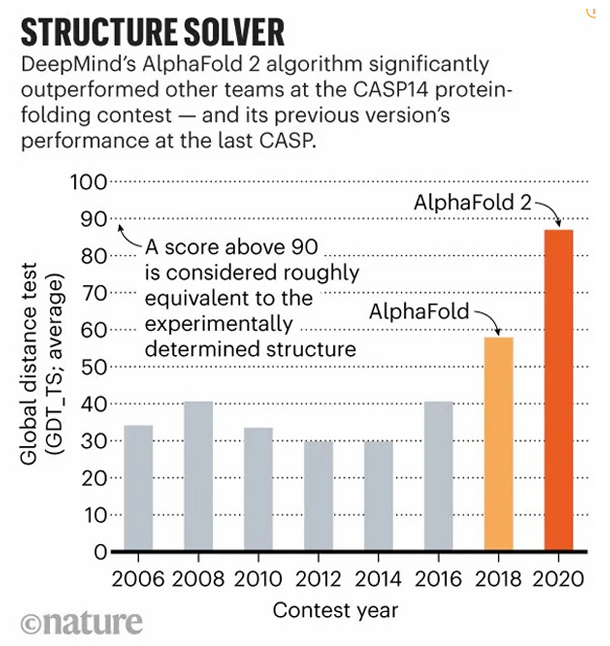
이후로 딥마인드 팀은 multimer 구조예측까지 가능한 AlphaFold Multimer, 리간드 및 핵산과 결합할 때 단백질의 역동적 구조변화를 예측하는 AlphaFold3를 2024년 5월 발표하여, 실제 생명체 내에서 단백질의 구조와 역동적 형태를 예측하는 방향으로 발전해왔다. 본 논문은 2같은 딥마인드 팀에서 AlphaFold2에 몇 가지 전략을 추가하여, missense variant의 pathogenicity를 예측하는 AlphaMissense를 만들고 이의 유용성을 평가한 논문으로 2023년 9월에 Science 지에 실린 논문이다. 여기까지만 들어도 화학자나 단백체 전문가가 아닌 필자가 얼마나 이를 이해하고 설명할 수 있을지 의문이 들었으나, 노벨상위원회에 따르면 190개국 200만명 이상이 이미 알파폴드를 사용하고 있다고 하니, 왠지 그렇게 어렵지 않을 것 같다는 생각으로 접근해보았다.
AlphaFold2의 핵심은 MSA (multiple sequence alignment)로, 여러 생물체에서 유래한 아미노산 서열을 정렬하여 공통된 패턴, 즉 진화적으로 잘 보존된 잔기 (residue)를 식별하는 기술이다. 이를 통해 어떤 서열이 기능적으로 중요하여 보존되는지 진화적 제약 (evolutionary constraint)을 파악하고, 단백질 입체 구조를 이해하는 것이 AlphaFold2의 기술이다. AlphaMissense는 이 MSA와 단백질 구조에 대한 이해를 토대로, missense variant의 병원성 (pathogenicity)을 예측하는 알고리즘이다. 원리를 요약하면 다음 그림과 같다.
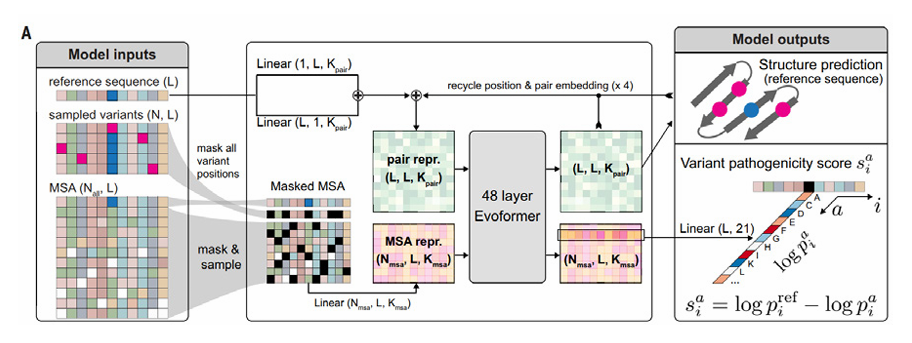
먼저 모델에 256개 잔기를 가진 참조 서열 (reference sequence)에 대해서, 다양한 변이를 가진 훈련 세트 최대 50개와, 다양한 종의 생물체에서 유래한 최대 2048개의 MSA를 입력값으로 넣는다. 이후 추론 과정에서 reference sequence를 첫번째 행으로, 모든 변이가 가려진 sampled variants를 두번째 행으로, 그리고 추가로 가려진 서열을 나머지 행으로 놓고 변이 위치를 마스킹한 MSA 훈련 세트에서 추론 과정을 거치게 한다. 마스킹이란 것은 아미노산 서열에서 변이가 발생한 위치를, 참조 서열의 해당 위치에서 숨긴다는 뜻이며, 이는 모델이 변이 위치의 정확한 정보를 직접 보지 못하도록 하여 변이의 병원성 예측 능력을 강화시키는 것이다. 이는 변이가 일어난 위치의 정보 없이도 주변 서열과 진화적 관계를 기반으로 예측하게 한다.
이후 AlphaFold2와 유사한 메커니즘으로 reference sequence에서부터 두 잔기 간 상호작용 정보인 pair representation을 받는데, 이는 단백질이 접히는 구조에 대한 이해를 돕는다. 또, 마스킹된 MSA로부터 MSA representation을 받는데, 이는 변이가 발생한 위치를 마스킹하여 직접적인 정보에 의존하지 않도록 하고, 주변 서열 및 진화적 맥락에서 병원성을 평가하도록 한다. 이후 pair representation과 MSA representation은 48층의 Evoformer 계층에 입력되어 정보가 결합되는 recycling 과정을 거친다.
이렇게 만들어진 모델을 토대로 참조 서열의 구조를 예측하고, 변이의 병원성 점수를 스칼라 값으로 계산해서 보여주게 된다. 병원성 점수는 참조 잔기 (reference residue)와 변이 잔기 각각의 log likelihood의 차이로 계산되며, 변이가 단백질 서열, 구조, 기능에 미치는 영향을 수치화한 것이다. 이렇게 계산된 병원성 점수에 대해 인간 또는 영장류에서 자주 관찰되는 양성 변이와, 관찰되지 않은 병원성 변이로 분류하여 미세조정 과정을 거친다. 양성 변이의 경우 MAF (minor allele frequency)에 따라 군집으로 분류하고, 희귀한 변이가 학습에 충분에 기여할 수 있도록 가중치를 더 주었다. 이는 희귀한 변이가 관찰 빈도가 낮아 데이터셋에서 상대적으로 부족하여, 모델이 고빈도 변이에 과적합(overfitting)되는 문제를 해결하고자 하였다. 원리에 대한 설명이 길었는데, 이후 내용은 대부분 딥마인드 팀의 AlphaMissense 자랑이다.
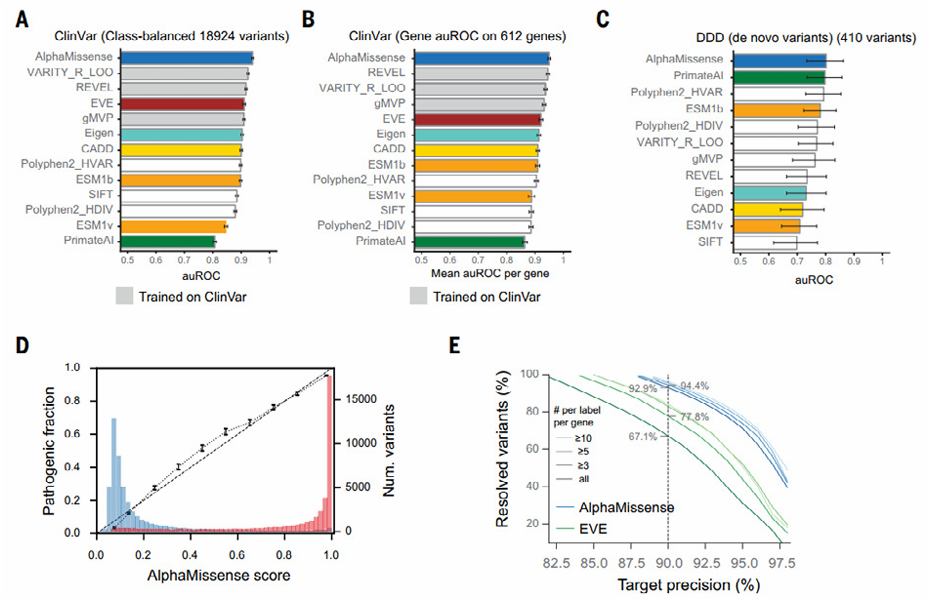
위 그림은 A) ClinVar의 999개 단백에서 9462개 병원성 및 9462개 양성 변이 구분, B) 유전자당 최소 5개 병원성 및 5개 양성 변이가 있는 612개 유전자, C) de novo variant cohort에서 215개 유전자의 353개 환자 및 57개 대조군 변이에서 다른 벤치마크 대비 우수했다는 이야기이다. D)는 ClinVar 82,872개 변이에서 병원성 점수의 분포를 보여주는데 빨간색 병원성 변이와 파란색 양성 변이를 잘 분류했다는 내용이다. E는 AlphaMissense와 유사한 원리로 구상된 EVE와 비교한 것으로 (그래서 A, B에서 빨간색 바 로 표시), ClinVar variant에 대해 다양한 정밀도에서 해결된 변이의 비율을 비교하여 EVE의 67.1%에 비해 92.9%로 크게 향상되었다는 자료이다.
뒤의 figure들도 대부분 비슷한 종류의 자랑이고, 본 기고의 목적은 AlphaMissense의 원리와 알고리즘을 알기 위한 것이었으므로, 이만 논문 요약을 줄이고자 한다 (혹시 궁금하면 본 논문을 chatGPT에 넣고 하나씩 물어보는 것도 추천한다). 혹시 좀더 내용이 궁금하다면 symoon9@gmail.com 혹은 오프라인으로 이야기를 해보아도 좋을 것이다.
