연세의대 진단검사의학과
크리스퍼 (CRISPR, Clustered Regularly Interspersed Short Palindromic Repeats)는 원래 세균 유전체에서 발견된 독특한 반복 서열로, 주로 침입한 파지(virus)의 염기서열과 유사한 ‘spacer’를 포함하고 있다. 이 서열이 전사되어 생성된 crRNA가 인접 유전자에서 발현되는 Cas 효소와 결합해 외래 DNA를 절단한다는 사실이 밝혀지면서, 이는 세균의 적응 면역 체계 중 하나로 이해되었다 [1].
이후 Jennifer Doudna와 Emmanuelle Charpentier는 crRNA의 서열을 교체하면 원하는 표적을 절단하는 ‘유전자 가위’로 사용할 수 있음을 제시하였고, 이 공로로 2020년 노벨 화학상을 수상하였다 [2]. 초기 CRISPR-Cas9 시스템은 표적 DNA에 존재하는 PAM(protospacer adjacent motif, 대표적으로 5’-NGG-3’) 인식 후 이중가닥을 절단하는 방식으로 작동하며, 이를 이용한 유전자 knockout 연구가 폭발적으로 확산되었다.
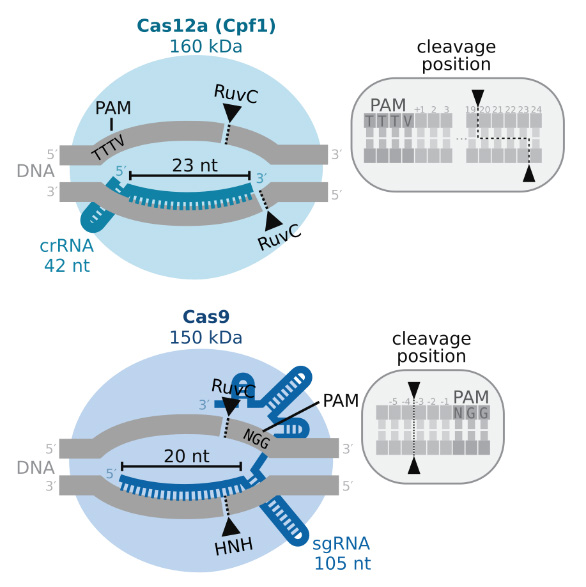 그림1
그림1
Cas9과 Cas12a 작용
이후 Cas9의 구조와 작동 기전이 정교하게 규명되면서 nickase Cas9을 기반으로 한 염기편집기(Base Editor)와 역전사효소를 결합한 프라임편집기(Prime Editor) 등 더욱 정밀한 편집 도구들이 개발되었다. Streptococcus pneumoniae에서 발견된 SpCas9이 가장 널리 쓰이는 반면, Cas12a는 ‘sticky end’ 형태의 절단을 만들어내는 특성을 지니며, Cas13은 RNA를 표적으로 한다는 점에서 독자적인 활용 영역을 가진다 (그림 1).
대규모 유전자 편집 연구의 확장
유전자 편집 기술의 고도화를 위해 다양한 연구가 진행되어 왔다.
대표적으로
◆ PAM 제약을 줄인 PAM-less 혹은 PAM-flexible Cas 개발
◆ 수만 종의 guide RNA를 기반으로 한 gRNA 효율 예측 모델 개발
◆ 오프타깃 반응을 최소화하기 위한 Cas 단백질 엔지니어링
◆ Cas 활성 및 편집 효율을 추정하는 AI 기반 모델링 등
이러한 연구는 대부분 대규모 pooled 실험을 통해 축적한 데이터를 기반으로 하며, 이는 CRISPR 기술을 연구 도구에서 치료 기술로 확장하는 핵심 기반이 되고 있다.
프라임편집기의 경우 특히 모든 형태의 염기 치환이 가능하고 정확도가 높아, 최적의 pegRNA를 설계하기 위한 PRIDICT, DeepPrime 등 다양한 예측 알고리즘이 제시되었다. 프라임편집기 자체도 PE1~PE7까지 지속적으로 개량되었으며, 예를 들어 MLH1 동역학을 조절하는 모듈을 결합한 PE4, La binding domain을 추가하여 편집 효율을 대폭 향상시킨 PE7 등이 대표적이다.
유전자 편집 기술은 유전자 치료제 개발뿐 아니라 기초 의생명 연구와 임상 유전학 분야에서도 매우 중요하게 활용되고 있다. 대표적 예가 CRISPRa/CRISPRi 시스템을 이용한 유전자 기능 연구이다. 절단 활성이 없는 dead Cas9(dCas9)에 전사 억제 혹은 활성화 도메인을 부착하고, 특정 유전자들을 표적하는 대규모 guide RNA 라이브러리를 도입하면, 약물 반응성·세포 생존·표현형 변화와 연관된 유전자를 효율적으로 스크리닝할 수 있다.
특히 진단검사의학 분야에서는 유전자 변이의 기능을 직접 측정하는 연구가 임상적 가치가 크다. 실제로 수천 가지 변이를 세포 수준에서 동시에 생성하고, 각 변이가 세포 생존이나 특정 분자 지표에 미치는 영향을 NGS 기반으로 정량화하는 Saturation Genome Editing(SGE)이 주목받고 있다. 이는 임상 유전학에서 해결이 어려웠던 불확실한 임상적 의의(Variants of Uncertain Significance, VUS)를 기능적으로 재분류할 수 있는 강력한 도구가 된다. 기본 원리는 ‘유전자 기능을 심각하게 떨어뜨리는 변이를 지닌 세포는 그렇지 않은 세포에 비해 선택 압력 하에서 생존이 불리하다’는 점을 이용하는 것이다. 또 다르게는 세포 내 특정 신호 경로의 활성, 전사체 변화, 수용체 변화 등 분자 바이오마커를 readout으로 삼아 변이의 기능적 영향을 정량화하기도 한다. 최종적으로는 NGS를 통해 각 변이의 비율 변화를 측정하고 통계적 모델링을 통해 non-functional, hypomorphic, functional 등으로 분류한다 (그림 2).
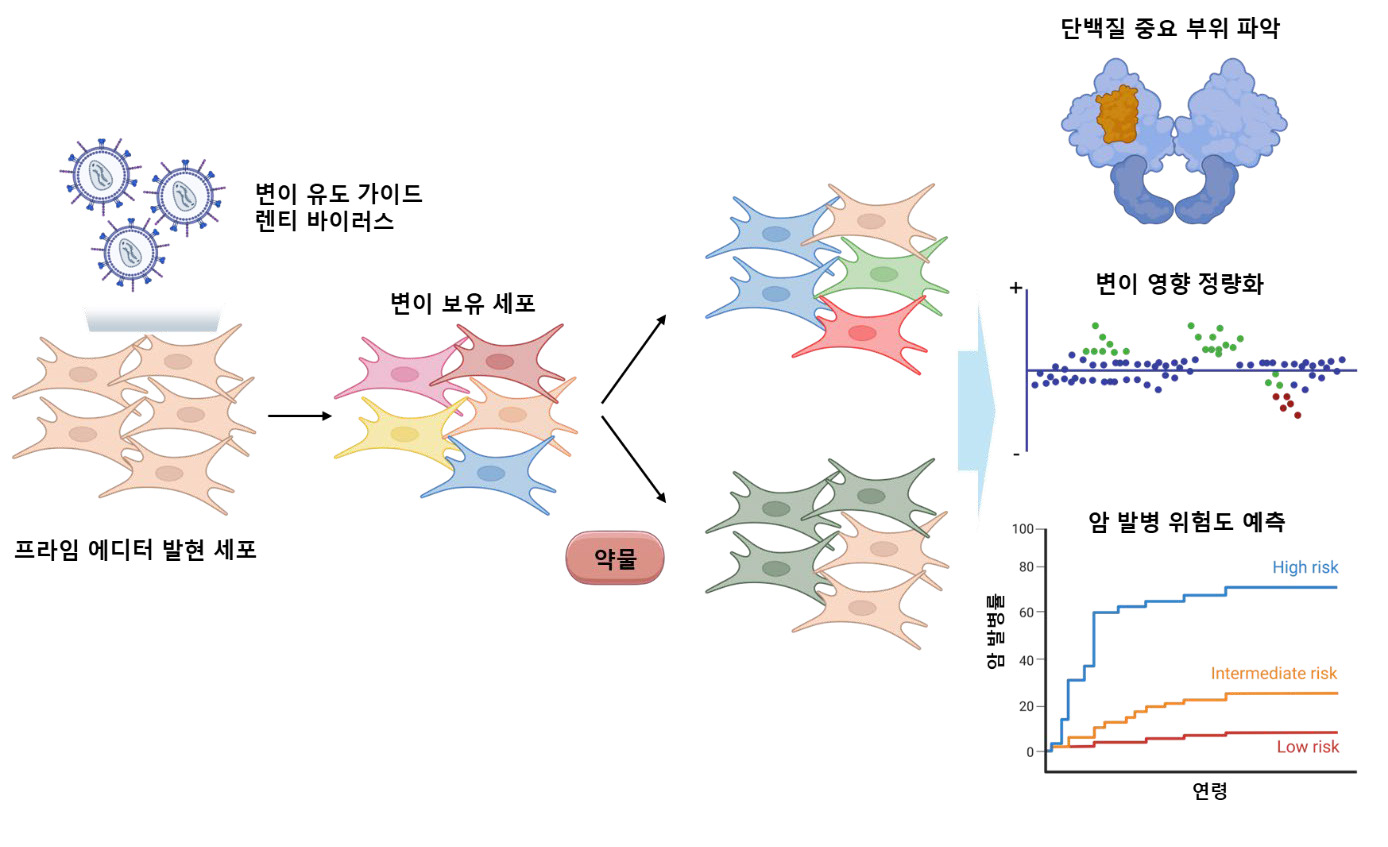 그림2
그림2
Saturation genome editing 실험의 개요
SGE 및 관련 기능 연구를 수행하기 위해서는 다음 요소의 선택이 매우 중요하다 (표 1).
| 단계 | 내용 |
|---|---|
| 적절한 세포주와 유전자 편집기 선택 | Cas9-HDR 방법: 표적 위치의 guide RNA와 함께, 표적 위치에 상동성을 가지며 원하는 변이를 포함한 oligonucleotide를 전달해야 함(그림 3). |
| 프라임편집 방법: 프라임편집 guide RNA를 전달해야 하며, 이는 표적 서열, primer-binding sequence, 변이를 포함하는 reverse-transcription template로 구성됨. | |
| 라이브러리 전달 | 변이 유도 guide RNA 라이브러리는 벡터 또는 바이러스 형태로 전달 가능함. |
| 세포 표현형 탐색 | 세포 성장 차이에 영향을 주는 유전자 변이는 NGS 변이 비율 차이를 통해 스크리닝 가능하며, 세포 염색 후 FACS 등을 통해 표현형을 분류하여 변이 분율 차이를 확인할 수 있음. |
| 정확성 검증 | All of Us, ClinVar, gnomAD, UK Biobank 등의 인구 데이터로 변이 분류의 정확성을 검증함. |
- – Cas9과 HDR 기반의 교정 방식이 널리 사용되지만, 유전자의 특성에 따라 Base editor, Prime editor가 더 적합할 수 있다.
- – 오프타깃 감소, 편집 정밀도 향상 등 목적에 따라 엔지니어링된 Cas 변형체 선택이 필요하다.
2. 세포주 선택
- – 표적 유전자의 발현량– 유전자 카피 수(LOH 여부)
- – 해당 pathway 내 다른 유전자 변이
- – 세포의 특정 환경 의존성(핵심 유전자 essentiality)
이는 편집 후 변이의 기능 차이를 얼마나 민감하게 구분할 수 있는지를 결정한다.
3. 표현형 판별 시스템 개발- – 생존 기반 스크리닝
- – 단백질 발현량/활성 변화 측정
- – 특정 신호전달 pathway의 리포터 시스템
이러한 측정법의 정밀도는 변이 기능 분류의 정확도를 좌우한다.
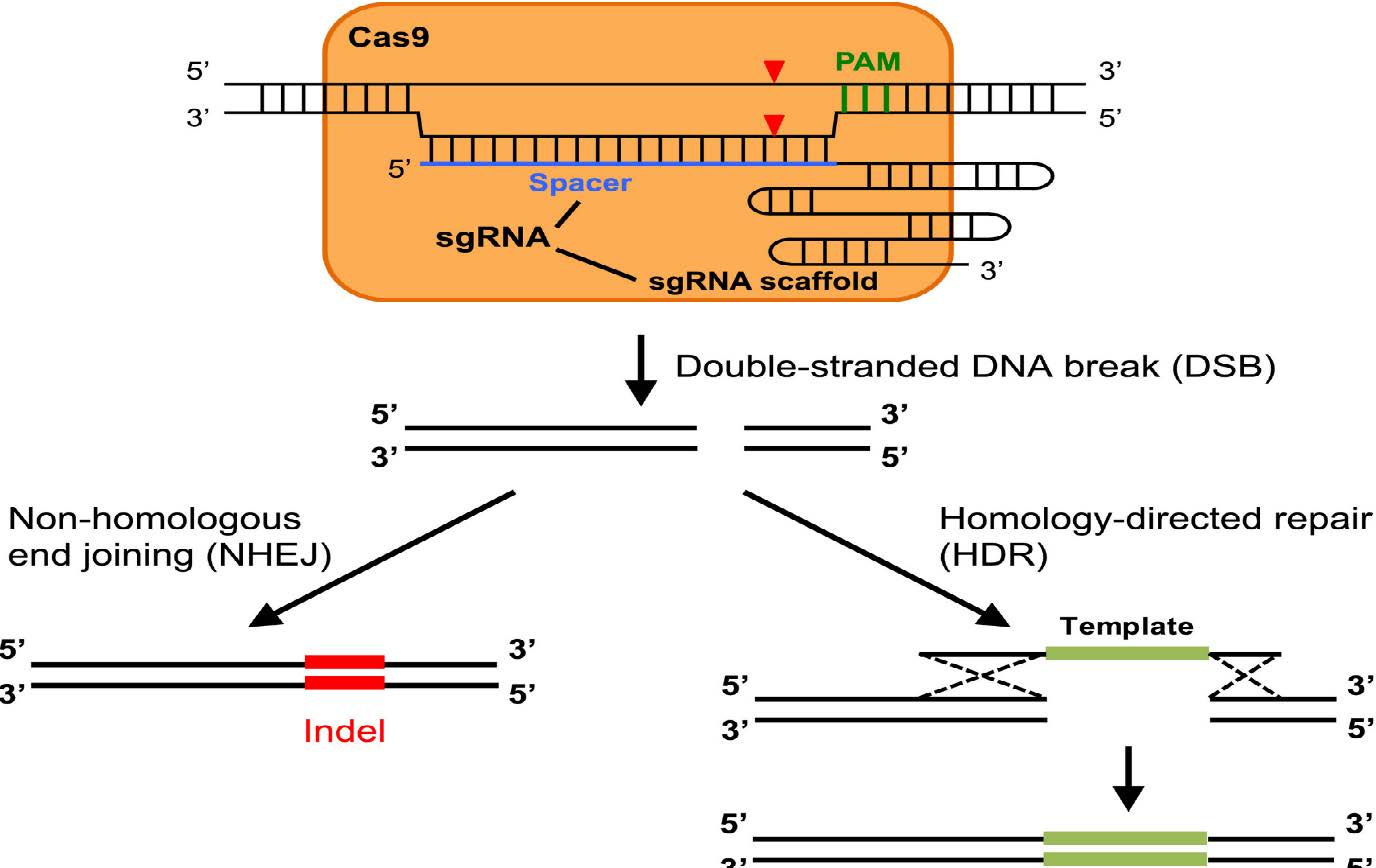 그림3
그림3
Cas9-HDR 시스템의 개요. Homology를 가지는 서열과 함께 원하는 변이를 함께 가지는 DNA template를 같이 주입하면 원하는 변이를 가지는 유전자 편집이 가능함.
(Shan S, Saoltis PS, Soltis DE, Yang B. Considerations in adapting CRISPR/Cas9 in nongenetic model plant systems. Appl Plant Sci. 2020;8(1):e11314. Published 2020 Jan 12. doi:10.1002/aps3.11314)
현재까지 BRCA1, BRCA2, ATM, TP53, RAD51C, MSH6, DDX4X, CHEK2, CDK12, LDLR, VHL 등이 다양하게 유전자 편집 기술을 이용하여 연구되었다. SGE의 대표적인 연구인 Findlay의 BRCA1 연구는 Cas9-HDR 방식을 이용하여 functional domain을 구성하는 13개 exon에 대해 single-nucleotide variant를 전수 조사하였으며 약 4,000개의 변이에 대한 function score를 도출해냈다 [3]. 이후 2024년 Mayo Clinic의 Fergus Couch 그룹이 BRCA2에 대한 동일 방식의 연구를 발표하며 기존의 evidence들과 결합하여 수천 개의 VUS를 재판별하였다 [4]. 이러한 연구는 영국에서 가장 활발히 진행되고 있으며 고해상도 기능지도(high-resolution functional map)가 여러 그룹을 통해 구축되고 있다.
개개의 변이를 평가했던 전통적인 기능연구와 다르지만, 그 정확도는 대부분 95-99%에 달한다. 하지만, 연구팀마다 편집 시스템과 readout을 분석하는 통계적 방법이 다르기 때문에 결과를 해석하는 데 있어서 주의를 요한다. 검사실에서는 동일 변이에 대한 여러 연구의 일치도, 수행된 연구의 신뢰도(변이가 높은 비율로 유도되었는지 여부, 라이브러리 구성의 적절성, 적절한 참조 서열을 가지고 있는 세포주의 사용) 등을 종합적으로 판단하여 적용해야 한다. SGE 결과만으로 판정을 내리기 보다는, 유전역학(가족력, segregation) 등을 모두 고려하여 최종 판정을 내릴 수 있도록 해야 하며, 보고된 적이 없는 변이의 경우 추가 검사에 대한 결정을 SGE 결과를 참조하여 판단할 수도 있겠다.
2025년도에는 이미 이러한 대용량 기능 연구들을 어떻게 가이드라인과 임상 해석에 접목할 것인가에 대한 논의가 시작되었다. 국내에서 검사 빈도가 높은 유전자들에 대해서 이러한 국제적인 논의에 참여할 수 있다면 향후 우리의 현실에 맞게 가이드라인을 수정, 보완하는데도 큰 도움이 될 것이다. 진단검사의학과 전문의는 NGS, 변이 해석뿐만 아니라, 실험의 원리와 한계, 통계적 방법에 대한 이해를 바탕으로 데이터의 신뢰도를 판단할 수 있어야 하며, ACMG 근거의 수준에 대한 주도적인 해석 능력을 필요로 할 것이다.
유전자 편집 기술은 이제 단순한 연구 도구를 넘어, 임상 유전학에서 변이의 기능적 해석을 정량적으로 제공하는 핵심 플랫폼으로 자리 잡고 있다. 특히 SGE와 같은 대규모 기능 분석은 임상 현장에서 가장 어려운 문제 중 하나였던 VUS 해석에 새로운 가능성을 제시하고 있으며, 향후 진단검사의학 분야에서 더욱 넓은 영역으로 활용이 확대될 것으로 기대된다.
[References]
1. Barrangou R, Fremaux C, Deveau H, et al. CRISPR provides acquired resistance against viruses in prokaryotes. Science. 2007;315(5819):1709-1712. doi:10.1126/science.1138140
2. Jinek M, Chylinski K, Fonfara I, Hauer M, Doudna JA, Charpentier E. A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science. 2012;337(6096):816-821. doi:10.1126/sci
ence.1225829
3. Findlay GM, Daza RM, Martin B, et al. Accurate classification of BRCA1 variants with saturation genome editing. Nature. 2018;562(7726):217-222. doi:10.1038/s41586-018-0461-z
4. Huang H, Hu C, Na J, et al. Functional evaluation and clinical classification of BRCA2 variants. Nature. 2025;638(8050):528-537. doi:10.1038/s41586-024-08388-8
