Notable Research
Functional assessment of all ATM SNVs using prime editing and deep learning
Technology Trend 기획이슈연세의대
ATM (ataxia telangiectasia mutated) 유전자는 63개의 엑손을 갖는 대형 유전자로, DNA 손상 반응(DNA damage response)의 핵심 조절자이자 종양 억제 유전자이다.
ATM 기능 상실은 다양한 암의 발생 위험을 증가시키고, 특히 유전성 유방암, 전립선암, 췌장암에서 예후에도 중요한 영향을 미친다.
그러나 현재까지 보고된 ATM 단일염기변이(SNV)의 상당수는 VUS(Variants of Uncertain Significance)로 분류되어 임상 해석이 어려운 상황이다.
특히, 유전성 암 증후군 선별 및 치료 반응 예측을 위해 ATM 변이의 기능적 평가가 필수적이지만, 유전자 크기가 커서 기능 분석이 기술적으로 난이도가 높다.
본 연구는 ATM의 가능한 모든 코딩 SNV(27,513개)와 인트론 SNV(1,442개)에 대한 기능적 영향을 체계적으로 규명하고자 하였으며,
이를 위해 프라임 에디팅 기반 대규모 세포 생존력 평가와 딥러닝 모델(DeepATM)을 결합하였다.
프라임 에디팅(Prime Editing) 기술은 Cas9 nickase와 역전사효소를 결합하여, 표적 변이 서열을 포함한 확장형 프라임 편집 가이드 RNA(epegRNA)를 통해 DNA 염기를 정밀 치환·삽입·삭제할 수 있는 최신 유전체 편집 기술이다.
연구진은 HCT116 세포에 ATM-haploid 모델을 제작하고, 렌티바이러스 기반 epegRNA 라이브러리를 도입하여 23,092개의 SNV를 실험적으로 평가하였다.
DNA 손상 반응을 유도하는 PARP 억제제인 Olaparib을 처리 조건에서 세포 생존율을 측정함으로써 ATM 기능 손실여부를 반영하는 지표 (기능 점수)를 산출하였으며, 실험적으로 기능 평가가 어려운 4,421개의 변이는 DeepATM을 통해 예측함으로써 전체 27,513개 변이에 대한 평가를 완성하였다.
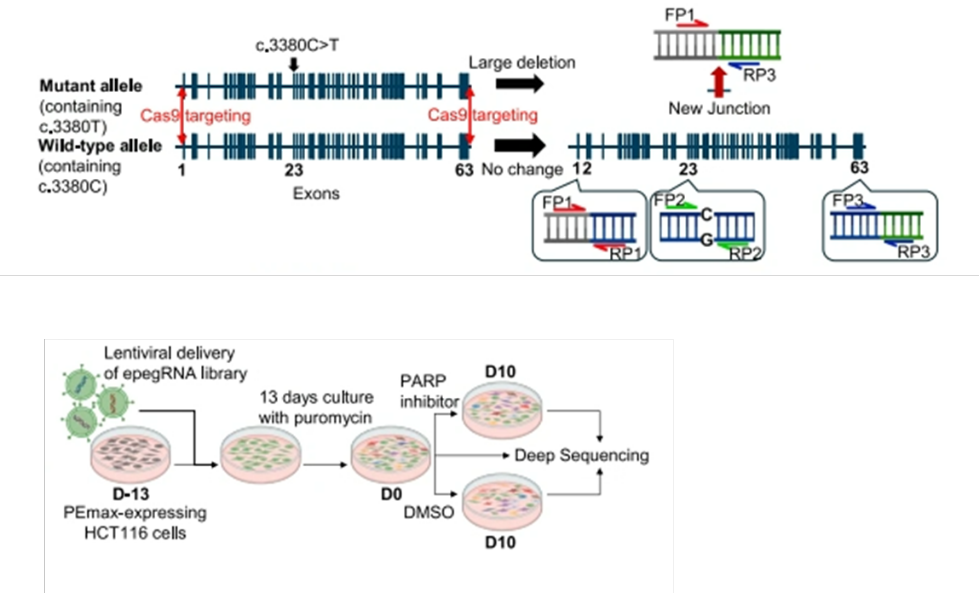 그림1
그림1
Cell line generation for functional evaluation of ATM variants 발췌 : HCT116 cell의 두개의 ATM 유전자 copy중 정상 copy만을 가진 ATM-haploid HCT116 세포 모델을 구축하였으며 제작된 epegRNA 라이브러리를 렌티바이러스 벡터에 탑재하여 ATM-haploid 세포에 도입하였다.
분석결과 대부분의 synonymous 변이는 중립적인 기능 점수를 보였으나 엑손-인트론 경계부위의 경우는 synonymous 변이임에도 일부의 경우 낮은 기능 점수를 나타냈다.
Nonsense 변이는 모든 엑손에 걸쳐 고르게 결핍되어 있었으며, 끝에서 두 번째 엑손(penultimate exon)과 3′ 말단 엑손에서도 마찬가지였다.
일반적으로 3′ 말단 엑손이나 끝에서 두 번째 엑손의 마지막 50개 뉴클레오타이드 내에서 발생한 nonsense 변이는 nonsense-mediated decay(NMD)를 회피할 수 있기 때문에, 이들 영역에서의 nonsense 변이 결핍은 해당 엑손들이 ATM 단백질 기능에 중요함을 시사하였다.
missense SNV의 경우, 엑손 57~60 (코딩 위치 8,269~8,786)에서 가장 강한 기능 손실이 관찰되었으며, 평균기능 점수는 각각 -2.1, -2.1, -3.6, -5.5 였다 (Figure 2A). 이 영역의 missense 변이 중 50% 이상이 비기능적(non-functional)이었으며, 나머지 엑손에서는 평균적으로 18%만이 비기능적이었다 (Figure 2C).
이는 해당 영역이 ATM 기능에 필수적임을 나타냈다.
이 영역은 고도로 보존된 kinase 도메인(엑손 55~63)에 포함되며, kinase 활성은 ATM의 DNA 손상 반응(DDR) 기능에 핵심적이다. ATM 단백질에 missense SNV에 대한 기능적 민감도를 매핑한 결과(Figure 2D), activation loop(잔기 2,888~2,911) 및 catalytic loop(잔기 2,867~2,875)에서 높은 민감성이 관찰되었다.
반면, 엑손 17(VUS가 자주 발견되는 엑손)에서는 396개의 missense 변이 중 단 13개(3.3%)만이 비기능적이었으며, 이는 이 영역이 ATM 단백질 기능에 덜 중요할 수 있음을 나타냈다.
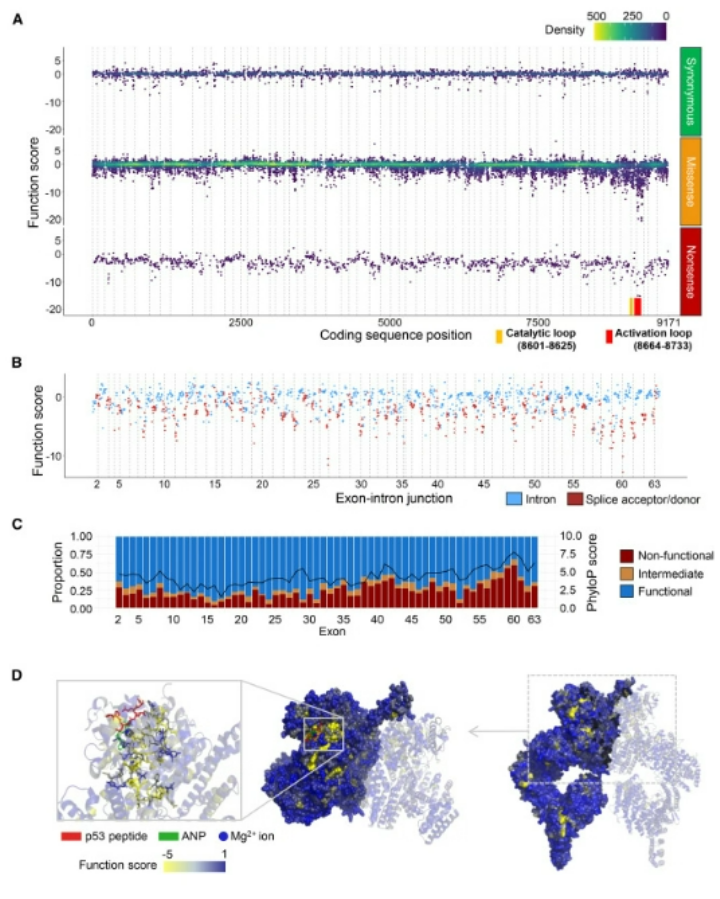 그림2
그림2
ATM 유전자 변이의 기능 점수 시각화: (A) 엑손 2-63 까지 SNV에 대한 기능 점수 (B) 인트론 및 수용체/공여체 (splice acceptor/donor) 변이에 대한 기능 점수 (C)엑손별 기능저하를 유발하는 missense 변이의 비율 (D) ATM 단백질의 3D 구조에 missense 변이에 대한 기능적 민감도
연구진은 실험 데이터를 기반으로 딥러닝 모델인 DeepATM을 개발하였다. 이 모델은 각 SNV의 염기 서열, 위치, 구조적 정보 등을 입력값으로 활용하여 기능 손실 여부를 예측한다. DeepATM은 기존의 예측 도구(CADD, REVEL, PolyPhen-2 등)와 비교하여 높은 정확도와 민감도를 보였으며, 특히 임상적으로 중요도가 높은 변이류에서 우수한 성능을 입증하였다. 모델의 성능은 ROC curve 및 precision-recall 분석을 통해 정량적으로 평가되었으며, 기존 임상데이터 베이스에서 평가가 되지 않은 변이에 대해 실험 데이터와 높은 상관관계를 보였다 (r=0.70). ClinVar 검증 세트에서는 병리적(P/LP) 변이와 양성(B/LB) 변이의 구분에서 AUC = 0.95, 특히 신뢰도 높은 ≥2-star 변이 세트에서는 AUC = 0.99로 매우 높은 분류 정확도를 보였다. 또한 AlphaMissense, EVE, ESM1b 등 최신 모델들과 비교했을 때도 DeepATM은 통계적으로 유의하게 높은 성능(p=0.034)을 나타냈으며, ATM kinase 도메인 외부의 변이 예측에서도 AUC = 0.99로 기존 모델을 능가하였다.
추가적으로, 연구진은 UK Biobank의 대규모 유전체-임상 데이터베이스를 활용하여 기능 손실 변이를 가진 개인의 암 발병률을 분석하였다. DeepATM 기반의 기능 저하 예측(experimentalized DeepATM, eDA)을 통해 비기능성 변이를 보유한 집단은 대조군에 비해 유방암, 전립선암, 췌장암 등의 발병 위험이 유의하게 증가한 것으로 나타났다 (Figure 3A). 예를 들어, UK Biobank 분석에서는 비기능성 변이 보유자에서 암 위험이 p =7.5 × 10-4 수준으로 통계적으로 유의하게 증가하였다. 또한 GENIE 암 데이터셋에서는 DeepATM이 예측한 비기능성 미평가 변이 698종에 대해 pan-cancer odds ratio (OR) = 52, p = 1.1 × 10-82 로 매우 강력한 연관성이 관찰되었다. 유방암 및 pan-cancer 위험 예측에서도 DeepATM은 기존 모델보다 일관되게 높은 OR을 보여주며, 실험 기반기능 평가의 임상적 타당성을 강하게 뒷받침하였다. 또한 암 환자 집단을 대상으로 한 생존 분석에서는 동일한 ATM 비기능성 변이가 암종에 따라 상반된 예후와 관련됨이 관찰되었다. 예를 들어, 만성 림프구성 백혈병 (CLL) 환자에서는 불량한 예후와, 진행성 방광암 환자에서는 상대적으로 좋은 예후와 관련되었다 (Figure 3B). 이러한 결과는 ATM 기능 평가가 단순히 발병 위험 예측에 그치지 않고, 암종별 환자 예후 stratification 에도 중요한 의미를 가질 수 있음을 시사하였다.
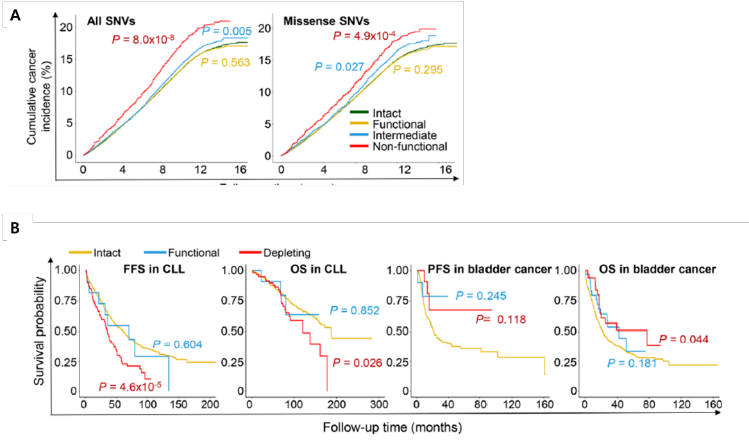 그림3
그림3
ATM 유전자 변이의 기능 점수와 임상 결과: (A)UK biobank 코호트에서 ATM 기능 손실 변이를 가진 집단의 누적 암 발생율이 대조군보다 높음 (B) Kaplan-Meier 분석에서 ATM 기능 손실 변이는 CLL에서는 불량한 예후, 진행성 방광암 환자에서는 양호한 예후와 연관됨.
본 연구는 ATM 유전자 내 모든 단일 염기 변이에 대해 프라임 에디팅과 약물 반응 분석을 통해 기능적 영향을 평가한 최초의 대규모 실험으로, 유전체 해석의 새로운 기준을 제시한다. 기존의 예측 중심 접근에서 벗어나 실제 생물학적 기능을 기반으로 한 평가를 수행함으로써, 임상 유전체 분석의 정확도와 신뢰도를 크게 향상시킬 수 있다. 특히 DeepATM 모델은 향후 유전체 기반 암 위험 예측, 진단, 치료 전략 수립에 있어 강력한 도구로 활용될 수 있으며, ATM 외의 다른 암 관련 유전자에 대한 유사한 접근이 기대된다. 본 연구는 정밀의학 시대에 실험–AI 결합 변이 해석 모델의 새로운 표준을 제시하는 중요한 이정표로 평가될 수 있다.
[References]
Lee et al., Functional assessment of all ATM SNVs using prime editing and deep learning, Cell (2025),
https://doi.org/10.1016/j.cell.2025.05.046
