씨젠의료재단
Clinical NGS의 검사 표준화는 언제쯤 실행될 수 있을까? clinical NGS에서 데이터 처리 과정은 환자의 진단 및 치료에 영향을 줄 수 있는 중요한 단계이나, 너무나 빠른 분석 파이프라인의 발전으로 인해 표준화와는 먼 방향으로 대세가 흘러온 것이 사실이다. 특히 우리나라에서는 식약처에서 clinical NGS와 관련된 검사 분야 별 지침서를 내놓는 등 전세계적으로 봐도 유래가 없을 정도로 많은 표준화 노력을 기울이고 있으나, 아직도 파이프라인과 변이 분석 방식, 그리고 데이터 통합 방식 면에서 많은 협의가 필요한 상태다. 변화하는 임상적 필요성과 데이터베이스, 분석 방식의 발전과 맞춰 표준화 방식도 변화가 필요한 시점이다. 이 글을 통해 NGS 표준화와 관련된 문제점을 알아보고, 적절한 해결책을 알아보기로 하자.
표준화와 관련하여서 검체 수집, 처리, 시퀀싱, 데이터 분석, 정도관리(quality control, QC), 데이터 스토리지 및 전송 등의 정책과 관련하여 다양한 요소들이 논의될 필요가 있다. 이번에 모든 관련 사항을 한꺼번에 논의하는 것은 지면 관계상 불가능할 것 같고, 시급한 몇 가지 문제, 그 중에서 참조 서열(reference sequence), NGS 성능 평가, 그리고 데이터 공유와 관련된 표준화라는 구체적인 주제에 대해서만 언급하고자 한다. CNV의 티어를 분류할 때 개별 환자의 임상 증상과 분리하여 판단해서 한 종류의 복제수변이는 각 환자의 임상적 중요성과 관계없이 일관된 해석이 되어야 한다.
1. 참조 서열(reference sequence)의 선정 NGS 검사가 사람을 대상으로 하는 경우 일반적으로 그 분석을 위해 reference sequence를 선정해야 한다. Reference sequence는 유전자 단편 정보의 위치를 추정하는 일종의 지도로써 사용된다 (그림 1) 이 때 우리는 이 지도를 hg19 (혹은 GRCh37)를 쓸지 hg38 (혹은 GRCh38)을 쓸지 선택을 해야 한다. 최근에 T2T (Telomere to Telomere)의 CHM13이더 정확한 맵핑(mapping) 결과를 얻을 수 있다는 연구가 등장하고 있으나, 면역세포 시퀀싱 등 일부 특수한 어플리케이션이 아닌 이상 당장 적용해야 할지 여부는 좀 더 지켜봐도 괜찮을 것 같은 상태다. 현실적인 범위 내에서 희귀질환이나 고형암 등에 적용 가능한 범위로 한정한다면, hg19나 hg38 중 하나를 선택하게 된다. hg38은 hg19보다 더 최신 버전으로 구조적으 로도 더 정확하고, 다수의 인구집단을 좀 더 잘 대변할 수 있다는 특성이 있다 [1]. 그럼에도 불구하고 많은 검사실에서 hg19를 쓰고 있다. hg19는 사소한 문제점 몇 가지를 제외하면 지금까지 비교적 잘 작동해왔고, hg38로 바꾸기에는 상당히 고통 스러운 과정을 거쳐야 한다. 기존에 hg19를 기준으로 보고한 병원 전산망에 남겨진 데이터들을 hg38로 변환하기 위해서는 genomic coordinate를 lift-over 시키거나, HGVS nomenclature로 표현된 DNA레벨의 서열이 hg38에서도 동일한 서열을 가지는지 확인해야 하는데, 전자도, 후자도 기술적으로 만만치 않다. 불가능하다는 말은 아니다. 실제 UCSC liftOver tool을 이용하면 ClinVar에 보고된 변이의 99% 이상을 정상적으로 hg19에서 hg38로 전환이 가능하다 [2]. 다만, 많은 병원들이 생물정보학 전문가의 지원 없이 NGS 검사를 수행하고 있고 의료의 보수적 특성까지 고려한다면 아마 목에 칼이 들어오는 그 순간까지 hg38로의 변환은 이루어지지 않을 것이다. 연구자에 따라서는 변환 문제가 그리 간단하지 않다고 보는 시각도 존재한다. 또한 hg38이 오히려 정확하지 않다는 비판도 있다 [3]. 예를 들어 21번 염색체에 잘못된 중복과 조립이 존재하며, 이로 인해 KCNE1, CBS, CRYAA, TRAPPC10, DNMT3L, KMT2C와 같은 임상적으로도 중요한 유전자 변이 호출에 영향을 준다는 것이다. 이러한 hg38의 미덥지 못한 부분을 고려한다면 hg19에서 hg38로 가는 과정은 건너뛰고 T2T project의 결과를 기다리는게 나을 수도 있다.
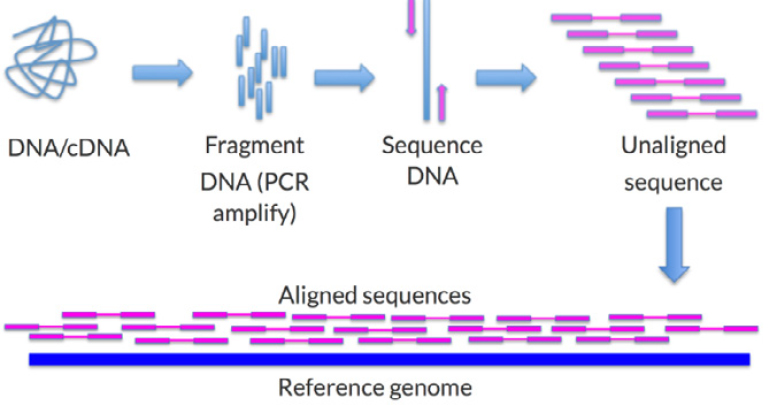 <그림 1> NGS의 reference 기반 mapping 방식 (출처: rockefelleruniversity.github.io)
<그림 1> NGS의 reference 기반 mapping 방식 (출처: rockefelleruniversity.github.io)
1.1 Clinically relevant transcript
일단 transcript 선정 문제부터 확인해보자. 이전에는 임상적으로 의미가 있는지 없는지 관계없이 longest transcript를 주로 써왔다. 이 방식은 coding 영역의 변이가 intron으로 잘못 오인되어 검사 대상에서 제외되는 문제를 줄여줄 수 있으 나, 완전히 방지하는 것은 불가능하며, 실제 임상적으로 연관성이 높지 않을 수 있다는 문제도 있다. 이를 해결하고자 유럽 생물정보학 연구소 (EMBL’s European Bioinformatics Institute, EMBL-EBI)와 미국 국립생물공학정보센터 (National Center for Biotechnology Information, NCBI)에서 각각 연구를 지속하다가 2022년 두 기관에서 MANE 및 MANE Plus transcript라는 새로운 표준을 제시하였다 [4]. 실제 임상적으로 의미가 있는 transcript를 기라성 같은 유럽과 미국의 중요 연구 기관 소속 유전학자분들이 지혜를 모아 만들었는데, 당연히 MANE이 이전 방식보다 훌륭하게 작동할 것이고, 의심할 여지없이 대세로 자리 잡을 것처럼 보인다. 그러나 불행하게도 hg19는 MANE transcript에 100% 호환되지 않는 다. MANE transcript는 현재 coding sequence는 hg19, hg38 모두 지원하긴 한다. 그런데 UTR 영역에서는 hg38만 지원되며, hg19의 경우 앞으로도 지원할 계획이 없다고 한다. UTR 영역의 임상적으로 중요한 영역으로 점차 인식되고 있음을 감안한다면 hg19를 계속 유지하기가 쉽지 않다고 생각된다. 현재 많은 기관에서도 사용 중인 유명 annotation tool 중하나인 Ensembl의 VEP (Ensembl Variant Effect Predictor)에서도 hg19를 쓸 경우 MANE transcript 표시가 제대로 지원되지 않으며 이 문제를 해결하기 위해서는 커스텀으로 파이프라인 수정 작업을 필요로 한다.
1.2 Reference sequence에 따른 데이터베이스 통합 문제
변이의 인구집단 중 비율을 표시하는데 쓰는 gnomAD 데이터베이스도 version(v)2까지만 hg19에 대응된다. v3부터는 hg38에 대해서만 매칭이 되며 현재는 v4까지 나온 상태이다. v4에서 방대한 양의 인구집단 데이터가 추가되었음을 고려한 다면, hg19를 선택했을 때 손해가 더 뼈아프게 다가온다.(그림 2)
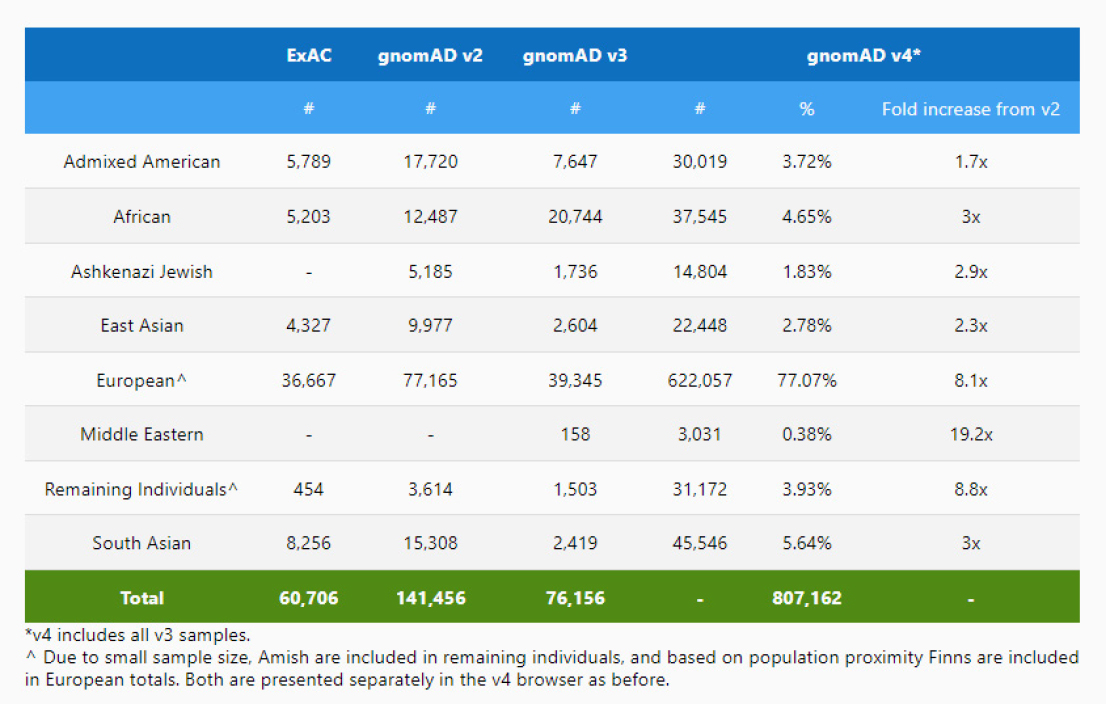 <그림 2>gnomAD 데이터베이스. hg19는 v2까지만 지원된다. v4와 v2는 검사한 사람 수 면에서 약 5.7배 차이난다. (출처: https://gnomad.broadinstitute.org/stats)
<그림 2>gnomAD 데이터베이스. hg19는 v2까지만 지원된다. v4와 v2는 검사한 사람 수 면에서 약 5.7배 차이난다. (출처: https://gnomad.broadinstitute.org/stats)
1.3 Reference sequence의 세부 버전 표준화
Reference sequence 문제와 관련하여 좀 다른 부분에서 접근해보자. 우리는 보통 우리가 hg19를 reference sequence 로 선정했다고 검사 결과지 하단에 적는다. 그런데 그게 진짜 동일한 hg19일까? hg19는 2009년에 등장한 이후 2013년까지 총 13번의 업그레이드가 이루어졌다. 그 뒤에도 일부 변형된 reference sequence가 등장했다. 예를 들면 hs37d5나 b37같은 것이 이에 해당한다. 이를 이용하여 매핑 할 경우 최신 hg19 버전인 hg19.p13보다도 정확한 변이 호출이 가능할수 있다고 보고된다.
hs37d5에서 가해진 대표적인 변형은 decoy sequence의 추가다. Decoy sequence는 오류를 일으키기 쉬운 염기서열을 미끼 서열에 정렬되도록 유도하여 변이 호출이 왜곡되는 것을 줄이는 방식으로 작동한다. 이를 이용해 많은 오류를 줄일 수있고 customizing도 얼마든지 가능하다. 그러나 기존 버전으로 정상적으로 검출되던 변이가 decoy version으로는 검출되지 않는 경우도 존재하기에 사용에 매우 주의해야 한다. 즉 우리가 결과지 하단에 적는 hg19라는 이름은 사실은 서로 조금씩 버전이 다를 수도 있고, 인간의 염기서열이 아닌 그 무언인가가 포함되어 있을 수 있으며, 심지어 특정 검사실에서만 쓰는 버전으로 업그레이드되어 사용되고 있을 수도 있다.
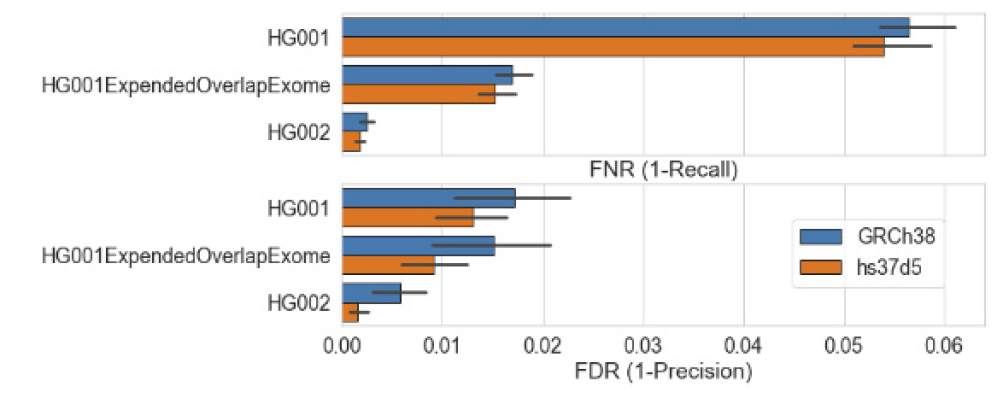 <그림 3>Reference sequence의 선택에 따른 성능 차이의 극단적인 예시. hg19의 variant 중 하나인 hs37d5는 hg38보다도 더 정확한 결과를 보여준다. 그림에서와 같이 false negative rate (FNR), false discovery rate (FDR) 면에서 더 우수한 결과를 보인다. hs37d5는 revised Cambridge Reference Sequence (rCRS)의 미토콘드리아 서열이나 인간 헤르페스 바이러스 서열에 대한 decoy sequence를 비롯한 더 정확한 변이 호출을 위한 변형이 가해졌다. 정통 hg19를 쓸지 동일 assembly 를 기반으로 하면서 표준 hg38보다 정확한 hs37d5을 쓸지 고민될 수밖에 없다. (출처: https://github.com/yihchii/blogpost_appendix/blob/master/Appendix_HG001_WES_sample_quality. md)
<그림 3>Reference sequence의 선택에 따른 성능 차이의 극단적인 예시. hg19의 variant 중 하나인 hs37d5는 hg38보다도 더 정확한 결과를 보여준다. 그림에서와 같이 false negative rate (FNR), false discovery rate (FDR) 면에서 더 우수한 결과를 보인다. hs37d5는 revised Cambridge Reference Sequence (rCRS)의 미토콘드리아 서열이나 인간 헤르페스 바이러스 서열에 대한 decoy sequence를 비롯한 더 정확한 변이 호출을 위한 변형이 가해졌다. 정통 hg19를 쓸지 동일 assembly 를 기반으로 하면서 표준 hg38보다 정확한 hs37d5을 쓸지 고민될 수밖에 없다. (출처: https://github.com/yihchii/blogpost_appendix/blob/master/Appendix_HG001_WES_sample_quality. md)
2. NGS 성능 평가 표준화 각 검사실마다 검체 처리 방식과 데이터 분석 방식에 따라 서로 천차만별의 검사 성능을 가질 수 있다. 그 중간 중간에 사용하는 장비, 시약, 분석 방식이 서로 다르기에 모두에게 동등한 방법으로 검사를 수행하라고 하는 것은 사실상 불가능하다. 이러한 상황에서 우리는 성능 평가를 통해 해당 시스템의 신뢰도를 평가하여 검사실 결과의 신뢰성에 대해 평가하곤 한다. 문제는 신뢰도 평가 자체가 사실 보통 일은 아니라는 것이다. 결과를 이미 알고 있는 검체에 대해 검사를 수행하고, 실제 결과가 일치 하는지 여부를 확인하곤 한다. 검체 당 알고 있는 변이의 수가 적은 경우 다양한 영역에 대해 성능 평가가 어렵기에 한 번에 여러 영역에서 변이를 확인할 수 있는 표준 물질, 특히 germline pipeline의 구성에 있어서는 전통적으로 표준물질 NA12878을 사용하는 편이다. NA12878과 같은 표준 물질에서 검출되는 변이의 숫자는 패널 구성에 따라 수 천개에 달할 수도 있다. 또한 평가하는 방식에 따라 같은 결과라 할지라도 그 검사의 정확도도 서로 다르게 평가될 수 있다. 예를 들어, NGS에서도 검출이 어려운 영역의 변이에 대해서도 평가를 시행할지 여부, 그것을 어떻게 결정할지에 대한 정책, 평가할 영역의 최소 coverage 기준치, multiple alternative allele에 대한 평가 정책 등 다양한 요소가 영향을 줄 수 있으며, 그 결정 사항에 따라 실제 검사 정확도가 5~10% 정도 차이가 날 수 있다. 미국 국립표준기술연구소 (National Institute of Standards and Technology, NIST)에서 Genome in a bottle (GIAB)라는 프로젝트 (https://www.nist.gov/programs-projects/genome-bottle)를 통해 검사 성능 표준화 방식을 연구하고 있다. 이 프로젝트에서는 NA12878을 비롯한 유대인 trio 데이 터에서 long read sequencing을 비롯한 다양한 시퀀싱 방법으로 얻은 데이터를 종합하여 이전보다 보다 정확한 염기서열 정보를 구성하였고, NGS 파이프라인에 대한 표준적인 평가 방법을 제시하고 있다. 실제 hap.py라는 파이썬 코드를 제공하고 있고, 이를 이용하면 복잡한 고려사항과 코딩 없이도 검사실 파이프라인의 정확도를 평가할 수 있다. 다만 성능평가 결과가 NGS 검사에 대한 평소 본인 생각보다 다소 좋지 않게 나올 수 있다. 실제, high-confidence region이라는 영역 내에서만 germline NGS 검사의 성능을 평가하기를 GIAB에서는 권장하고 있는데 평가하고자 하는 패널에 NGS 검출에 까다로운 영역이 다수 포함된 경우 이 권고안을 무시하면 상당히 낮은 성능 평가 결과를 보게 될 것이다.
GIAB 프로젝트는 검사 운영과 파이프라인의 세부 조정에 큰 도움을 줄 수 있다. 예를 들어 적정한 수준의 검사 정확성을 보장 하기 위한 평균 coverage를 산출한다거나, GATK 하드 필터링 기준을 평가할 때 유용하다. 또한 타 검사실 간 비교 시에도 보다 공정한 성능평가 결과를 보장해 줄 수 있을 것이다.
3. 검사실 간 NGS 데이터 공유 최근 유전체 정보, EMR (electronic medical record) 정보와의 통합, 클라우드 시스템 구축 등 유전체 분석 관련 보건의료 데이터 통합에 여러 연구가 도전하고 있다. 아울러 기존에 수행한 NGS 검사 결과에 대한 재분석 요구는 날로 높아지고 있다. 기존 분석한 NGS 검사 결과에 대해 재현 및 교환 가능한 형태로 데이터를 저장하고 전송하기 위해서는 어떠한 요소가 결정되어야 할까?
3.1 공유 파일 형식에 대한 표준화
만약 A 병원의 환자가 시행한 NGS 데이터를 B 병원에서 다시 분석하고자 할 때, 병원 간 NGS 데이터 공유를 위해서는 몇 가지 고려해야 할 사안이 존재할 것이다. 먼저 데이터를 전송할 때 어떠한 형태로 보내는 것이 좋을지 생각해보자.
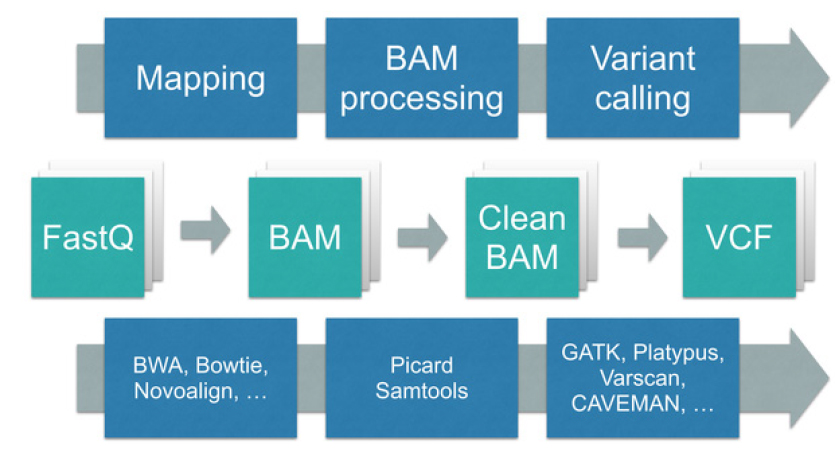 <그림 4> 분석 어플리케이션에 따라 큰 차이는 있으나, FASTQ, BAM, VCF는 대체로 분석 파이프라인 상 공통 분모에 해당한다. 어느 파일을 공유하는 게 편할까? (출처: PLoS One. 2015 Jul 16;10(7):e0132868.)
<그림 4> 분석 어플리케이션에 따라 큰 차이는 있으나, FASTQ, BAM, VCF는 대체로 분석 파이프라인 상 공통 분모에 해당한다. 어느 파일을 공유하는 게 편할까? (출처: PLoS One. 2015 Jul 16;10(7):e0132868.)
| 파일형태 | 장점 | 단점 |
|---|---|---|
| FASTQ | 높은 데이터 호환성 | Mapping 단계부터 processing 필요 |
| 저장 공간 문제 발생 | ||
| BAM | Mapping 단계 생략 가능 | Reference가 다른 경우 데이터 손실 가능성 존재 |
| VCF | 전처리 단계 최소화 | Pipeline 오류가 있는 경우 영향 큼 |
파일 공유 시 BAM 파일과 FASTQ 파일을 같이 공유할 필요는 사실 그리 크지 않다. BAM과FASTQ는 서로 상호 전환이 가능하며, 파일 사이즈 면에서도 큰 차이는 없다. VCF 파일의 경우 FASTQ나 BAM에 비해 상대적으로 크기가 10배 이상 작으므로, VCF을 포함하여 공유하더라도 전체 데이터 용량에 미치는 영향은 크지 않다. 그러므로 VCF 파일은 항상 공유하고, 추가적으로 변이에 대한 evaluation 목적으로 BAM를 쓸 것인지, FASTQ를 쓸 것인지에 대한 문제가 남는다. Mapping 자체는 기관 마다 그 결과물 차이가 크지 않을 것으로 예상되지만, reference sequence의 차이는 서로 치명적인 차이를 만들 수 있기에, FASTQ 파일 레벨에서 공유하는 것이 좀 더 안전하다. 반면, 이 방법은 BAM파일까지 processing하는데 상당한 컴퓨팅 리소스를 소모하게 될 것이다. 그러므로 FASTQ를 BAM으로 전환 시 발생할 수 있는 정보 손실과 기관 간에 염기서열 mapping 방식의 차이의 정도를 조사할 필요가 있다. 구체적으로 각 기관마다 사용하는 reference sequence의 구체적인 버전과 decoy 서열에 매핑된 read를 최종 BAM파일에서 제거하는지 여부에 대한 정책, duplicated read의 제거 (de-duplication) 여부 등의 요소 등에 대해 조사할 필요가 있다. 만약 기관 간에 해당 방식이 서로 협의가 불가능한 수준의 분석 방식에 심각한 차이를 보인다면, 시간과 분석 자원이 더 든다 하더라도 FASTQ 파일 레벨에서 공유하는 것이 안전하다고 생각한다.
3.2 파일 전송과 관련된 문제
NGS 데이터는 그 크기가 커서 보관 및 전송에 어려운 점이 존재한다. 또한 그 과정 중에서 파일이 변형, 손상되는 일이 발생할 수 있어, 적절한 데이터 처리를 위해서는 MD5(Message-Digest algorithm 5)나 SHA256 (Secure Hash Algorithm 256) 해시(hash: 단방향 암호화기법) 알고리즘을 활용한 무결성 검증이 도움이 될 수 있다. 또한 의료 보안 문제와 관련하여 최근 미국에서 도입 중인 HIPPA (Health Insurance Portability and Accountability Act) 표준을 따를지 여부에 대해서도 결정할 필요가 있다.
 <그림 5> NGS 파이프라인의 input 데이터에 변형이 생기는 경우 다른 병원에서 파이프라인이 정상적으로 작동할 지에 대해서는 보장할 수 없다. USB나 외장 하드 등으로 병원 간 데이터를 전송하거나, 웹으로 전송하고자 할 때 대용량 파일 전송 중 파일 무결성을 확인하는 방법 중 하나가 MD5 hash를 확인하는 것이다. MD5 hash를 확인하는 툴을 통해 hash를 생산하고, 보내기 전에 작성한 hash와 보낸 후의 hash가 서로 일치해야 파일이 변형 및 손상되지 않았다고 판단할 수 있다. 이 과정은 데이터를 수신하고자 하는 의료 기관과 협의가 필요하다. (출처: https://blog.systoolsgroup.com/md5-algorithm-for-forensics/)
<그림 5> NGS 파이프라인의 input 데이터에 변형이 생기는 경우 다른 병원에서 파이프라인이 정상적으로 작동할 지에 대해서는 보장할 수 없다. USB나 외장 하드 등으로 병원 간 데이터를 전송하거나, 웹으로 전송하고자 할 때 대용량 파일 전송 중 파일 무결성을 확인하는 방법 중 하나가 MD5 hash를 확인하는 것이다. MD5 hash를 확인하는 툴을 통해 hash를 생산하고, 보내기 전에 작성한 hash와 보낸 후의 hash가 서로 일치해야 파일이 변형 및 손상되지 않았다고 판단할 수 있다. 이 과정은 데이터를 수신하고자 하는 의료 기관과 협의가 필요하다. (출처: https://blog.systoolsgroup.com/md5-algorithm-for-forensics/)
4. 표준화 없이 버티면 어떻게 될까? 솔직히 말하면 표준화 문제에 대한 솔직한 해결책을 말할 수 있는 사람은 많지 않다고 생각한다. 이번 주제에서 표준화 과정에 제시된 난제만 해도 쉬운 것이 없어 보인다. 그러나 표준화 없이 버티면 생기는 문제에 대해서는 확실히 말할 수 있을 것 같다. hg19의 사용을 지속하면 연동 가능한 최신 데이터베이스의 숫자가 점차 줄어들 것이며, transcript 선정에 어려움을 계속 느껴야 할 것이다. 또한, 성능 평가에 대한 표준화가 늦으면, 파이프라인 성능 개선에 기준을 잡지 못할 것이며, 마지막으로 유전체 데이터의 공유에 대한 표준 선정이 늦을수록 관련 연구 사업을 수행하는데 어려움을 느끼게 될 것이다.
표준화의 방식은 여러 가지가 존재할 수 있다. 사용하는 파이프라인과 reference sequence를 깐깐하게 다 지정해가며, 서로 이탈하지 않도록 종용하는 방식과, 적정 성능을 이룰 수만 있으면 그 어떠한 새로운 툴이나 방식도 허용하는 방식, 혹은 그중간도 가능할 것이다. 개인적으로 생각하는 가장 이상적인 방식은 느슨한 단계에서 표준화를 하되, 그 중 임상적으로 큰 영향을 미칠 수 있는 과정에 대해서는 서로 공유하고 같이 고민해보는 것이다.
NGS 검사는 불행히도 표준화와는 거리가 먼 분야로, 임상에 필요에 의해 급격하게 도입되었다. 이를 통해 우리는 유전 분야 에서 다른 국가에 비해 상대적으로 앞서 나갈 수 있었으며, 높은 검사 역량을 보유할 수 있었다. 이제는 그 대가를 치를 때다. 우리는 그 동안 작성한 레포트에 보고한 변이를 앞으로 어떠한 방법을 통해 변경된 유전체 지도에서 표현할 수 있을지, 앞으 로는 어떤 데이터를 추가해야 향후 발생하는 변화에 대해서도 유연하게 대처할 수 있을지 고민해야 한다. 또한 쌓여 있는 데이터를 어떻게 상호 교환이 가능한 형태로 전환할지 협의해야 할 것이다.
[References]
1. Guo Y, Dai Y, Yu H, Zhao S, Samuels DC, Shyr Y. Improvements and impacts of GRCh38 human reference on high throughput sequencing data analysis. Genomics. 2017 Mar;109(2):83-90. doi: 10.1016/j.ygeno.2017.01.005. Epub 2017 Jan 26. PMID: 28131802.
2. Luu PL, Ong PT, Dinh TP, Clark SJ. Benchmark study comparing liftover tools for genome conversion of epigenome sequencing data. NAR Genom Bioinform. 2020 Aug 6;2(3):lqaa054. doi: 10.1093/nargab/lqaa054. PMID: 33575605; PMCID: PMC7671393.
3. Behera S, LeFaive J, Orchard P, Mahmoud M, Paulin LF, Farek J, Soto DC, Parker SCJ, Smith AV, Dennis MY, Zook JM, Sedlazeck FJ. FixItFelix: improving genomic analysis by fixing reference errors. Genome Biol. 2023 Feb 21;24(1):31. doi: 10.1186/s13059-023-02863-7. PMID: 36810122; PMCID: PMC9942314.
4. Morales J, Pujar S, Loveland JE, Astashyn A, Bennett R, Berry A, Cox E, Davidson C, Ermolaeva O, Farrell CM, Fatima R, Gil L, Goldfarb T, Gonzalez JM, Haddad D, Hardy M, Hunt T, Jackson J, Joardar VS, Kay M, Kodali VK, McGarvey KM, McMahon A, Mudge JM, Murphy DN, Murphy MR, Rajput B, Rangwala SH, Riddick LD, Thibaud-Nissen F, Threadgold G, Vatsan AR, Wallin C, Webb D, Flicek P, Birney E, Pruitt KD, Frankish A, Cunningham F, Murphy TD. A joint NCBI and EMBL-EBI transcript set for clinical genomics and research. Nature. 2022 Apr;604(7905):310-315. doi: 10.1038/s41586-022-04558-8. Epub 2022 Apr 6. PMID: 35388217; PMCID: PMC9007741.
