Technology Trend
Long-read capture with Twist target
enrichment system
Technology Trend
Nortable Research
1. Introduction Targeted resequencing은 Whole genome sequencing분석보다 접근성이 높은 규모와 비용으로 유전자 영역의 high resolution분석을 가능하게 한다. long-read PacBio HiFi sequencing이 임상적으로 활용 가능한 복잡한 유전자 loci를 정확하고 포괄적으로 조사하는 것으로 나타났지만, 연구는 주로 PCR amplicon기반 방법을 사용하여 단일 유전자에 초점을 맞추었다. 여기에서는 HiFi read로 sequencing된 유전자 패널에 Twist Bioscience의 target enrichment workflow를 활용하는 방법을 소개한다. 이 포스터는 협업을 통해 개발된 2개의 alliance panel (50개 유전자로 구성된 약리유전체학 패널과 약 400개의 판독이 어렵고, 의학적으로 관련성이 있는 ‘dark genes’)의 특징과 성능을 보여준다.
2. Methods 본 연구에서는 0.2~22 MB의 다양한 표적 크기를 가진 유전자 패널을 설계하였다. probe는 전용 알고리즘을 사용하여 최적화되었으며 복잡한 영역을 균형 있게 capture하는 동시에 표적 외(off-target) 염기서열의 capture를 줄일 수 있다. Twist사의 long read hybrid capture protocol1에 따라, mechanical fragmentation 방법 (ex: Diagenode Megaruptor 또는 Covaris g-TUBE)를 사용하여 절단(shearing)된 200~1000ng로 단편화된 gDNA를 준비한다. End-repair 및 A-tailing하고 truncated Y-shaped adapters를 adapted gDNA에 결합 (ligation)한다. PCR 중에 sample barcoding을 위한 10bp (unique dual indices, UDI)을 첨가하였다. 4~8개의 sample은 overnight 동안 hybridization하기 위해 pooling하였다. Capture 후 라이브러리는 SMRTbell® prep kit 3.0을 사용하여 SMRTbell 라이브러리 준비를 수행하고 30시간 movie time으로 PacBio Sequel IIe에서 sequencing을 수행한다. Target 크기에 따라 최대 400개 sample을 5~10 kb의 8M HiFi read length로 한 SMRT Cell에서 multiplexing 및 sequencing을 수행할 수 있다.
 SMRT Link was used to generate HiFi reads, remove PCR duplicates, and demultiplex, and a PacBio WGS pipeline was used to call variants for individual
samples. The PacBio human WGS workflow is publicly available on
SMRT Link was used to generate HiFi reads, remove PCR duplicates, and demultiplex, and a PacBio WGS pipeline was used to call variants for individual
samples. The PacBio human WGS workflow is publicly available on
GitHub: https://github.com/PacificBiosciences/pb-human-wgs-workflow-snakemake
3. Long Read Pharmacogenomics (PGx) Panel HLA 유전자 및 CYP2D6을 비롯하여 약물에 반응하는 유전자는 low sequence complexity 또는 highly homologous pseudogenes을 가지고 있기 때문에 array나 short read sequencing을 통한 유전자형 분석이 어려운 것으로 알려져 있다. 또한, CYP2D6는 일반적으로 유전자 일부, 또는 전체 유전자의 duplications 및 rearrangements를 가지고 있기 때문에 마찬가지로 어렵다. 연구진은 50개 유전자 약물유전학 패널을 개발하기 위해 노력하였다. nuclear contents가 추가되어 전체 mitochondria genome을 커버하기 때문에 이종조직성(herteroplasmy)의 동시 검출이 가능하다.
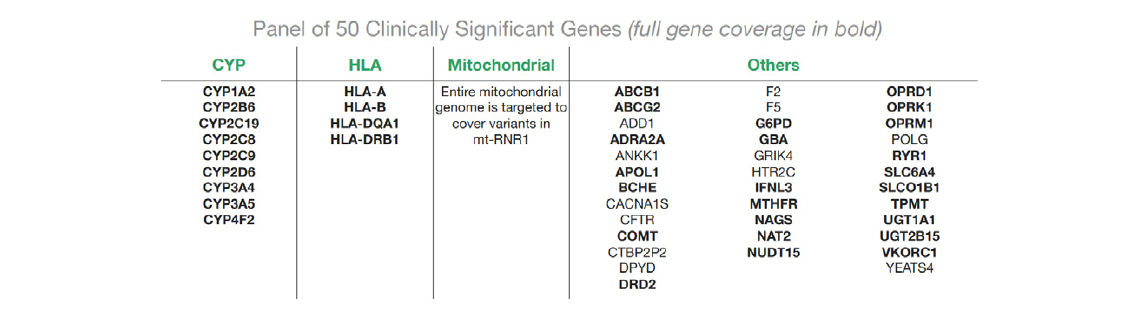
24개의 GET-RM Coriell sample에 대해 Sequel IIe system에서 1개의 SMRT Cell 8M을 통해 염기서열을 분석하였다. Sample은 평균 150k의 HiFi reads을 가지며, mean read length는 최대 5.3kb였다. 다운스트림 분석에서 duplicate의 2%만 제거되었다. 모든 target에서 190x mean target coverage가 확인되었다. 모든 sample에서 target 영역의 96%가 20x coverage를 초과했으며 93%가 30x coverage를 초과하였다.
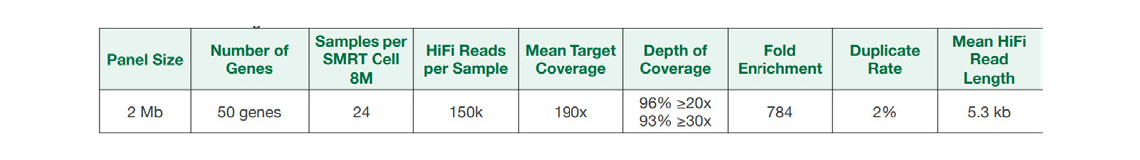
Short-read WGS data(bottom track)가 gap과 다양한 coverage를 보인 반면, Targeted regions (top track)의 coverage는 gap이 없이 균일성을 보였다.
 * Short-read WGS PE250:
* Short-read WGS PE250:
https://ftp-trace.ncbi.nlm.nih.gov/ReferenceSamples/giab/data/AshkenazimTrio/HG002_NA24385_son/NIST_Illumina_2x250bps/novoalign_bams/
4. Long-Read Dark Genes Panel 의학적으로 관련성이 있는 389개의 유전자가 갖는 반복적 특성과 다형성 복합도는 임상 환경에서 정확한 분석에 어려움이 있다. 하지만 HiFi assemblies2를 통해 최대 70%까지 해결할 수 있다. 이 패널은 " NGS dead zone’"에 있는 short-read로 sequencing이나 맵핑이 어려운 여러 유전자를 포함하여 염기서열 검출이 어려운 389개의 유전자와. 대한 full gene coverage를 제공한다3,4. 이러한 유전자는 심혈관 질환, 신경병증, 면역결핍, 시력 관련 질환 등 다양한 질환에 영향을 미치는 것으로 보고되고 있으며, 암 유발 유전자(예: PTEN)도 여기에 포함된다.

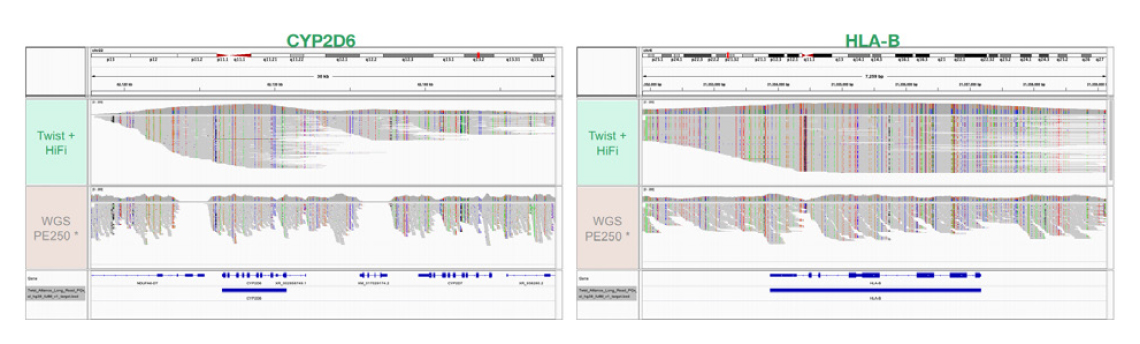
4개의 Coriell sample에 대해 Sequel IIe system에서 1개의 SMRT Cell 8M을 통해 염기서열을 분석하였다. Sample은 평균 893k의 HiFi reads을 가지며, mean read length는 최대 5.3kb였다. 다운스트림 분석에서 duplicate 중 3%만 제거되었다. 모든 target에서 mean target coverage 75x가 달성되었다. 모든 sample에서 target region의 93%가 10x coverage를 초과했으며 90%가 20x coverage를 초과하였다.
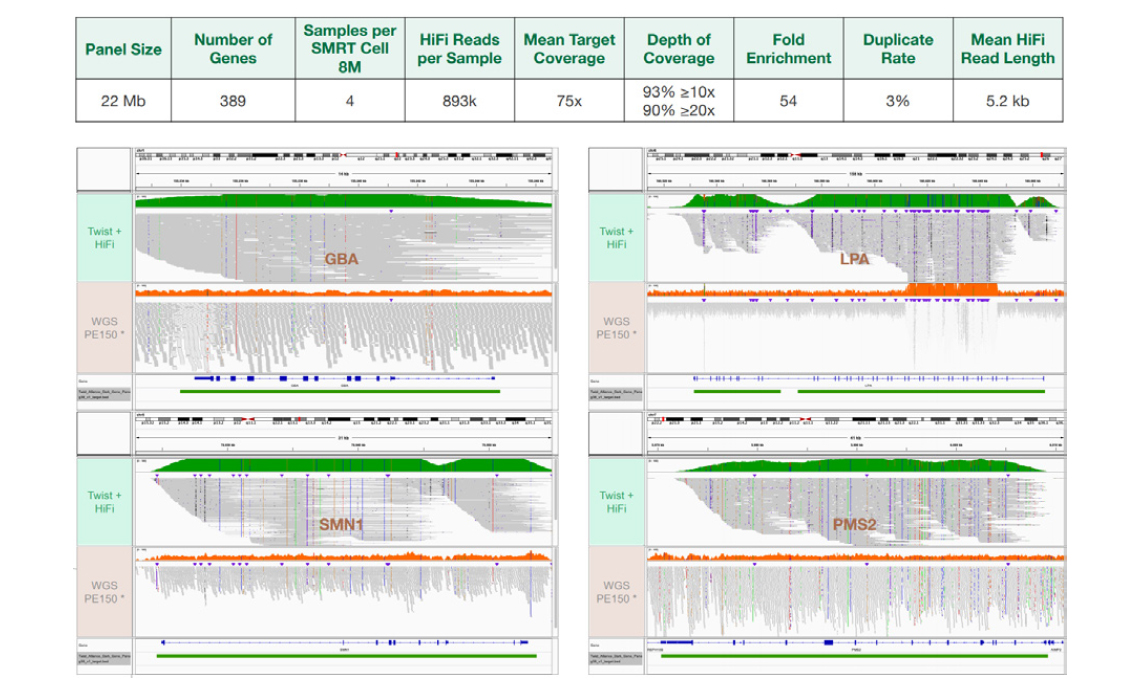 * Short-read WGS PE150:
* Short-read WGS PE150:
https://storage.googleapis.com/brain-genomics-public/research/sequencing/grch38/bam/novaseq/wgs_pcr_free/30x/HG001.novaseq.pcr-free.30x.dedup.grch38.bam
5. Conclusions 연구진은 Twist사의 long-read capture method가 다양한 크기의 multiple gene panel로 실행된 Coriell sample을 사용하여 효율적으로 유전자 표적의 포괄적인 커버리지를 가능하게 함을 입증하였다. 여기서 강조하는 것은 CYP2D6, HLA, SMN1, LPA와 같은 복잡한 영역입니다. 이 long-read capture protocol은 Twist custom 또는 fixed gene panel과 함께 long-read sequencing을 사용하여 관심 유전자를 효율적으로 capture할 수 있다. Contents(target 영역)를 추가하기 위해서는 hybridization 과정 중에 이차 패널(spike-in)을 쉽게 첨가할 수 있다. 입증된 방법을 통해 long read length로 확장이 가능하고 비용을 절감할 수 있는 hybrid capture를 가능하게 하여, coverage bias를 최소화하고, 정확도를 최대화하며 모든 변이 유형을 완벽하게 capture할 수 있다. 여기에는 short-read 및 Sanger sequencing으로 접근할 수 없는 structural variation과 haplotype phasing정보가 포함된다.
6. Reference
1. Twist Long Read Library Preparation and Standard Hyb v2 Enrichment
https://www.twistbioscience.com/resources/protocol/long-read-library-preparation-and-standard-hyb-v2-enrichment
2. Sedlazeck et al. Curated variation benchmarks for challenging medically relevant autosomal genes. Nature Biotech (2022)
3. Mandelker et al Navigating highly homologous genes in a molecular diagnostic setting: a resource for clinical next-generation sequencing.
Genetics in Medicine (2016)
4. Wenger et al. Accurate circular consensus long-read sequencing improves variant detection and assembly of a human genome. Nature Biotech (2019)
7. Acknowledgements The authors would like to thank Stuart A. Scott and his group at Stanford University for the content curation of Pharmacogenomics Panel and Fritz Sedlazeck and his group at Baylor College of Medicine Human Genome Sequencing Center (BCM-HGSC) for Dark Genes Panel. The authors would also like to thank the technical assistance of Sarah Kingan, Nina Gonzaludo, John Harting, Xiao Chen, Christine Lambert, and Primo Baybayan at PacBio.
* 원문자료는 아래 링크에서 다운로드 받으실 수 있습니다.
https://drive.google.com/file/d/1Sje2_am3LsBuf4kmb2ofgLIRLhU9VFlD/view?usp=sharing
