Nortable Research
Parallel sequencing of 87 STR and 294 SNP markers using the prototype of the SifaMPS panel on the MiSeq FGx™ system
사용자 경험 최신 보험정보씨젠의료재단
1990년대 상용화된 이후로 임상 혹은 법의학에서 개인식별검사는 모세관 전기 영동법(capillary electrophoresis, PCR-CE)을 이용한 단연쇄반복(Short tandem repeat, STR) 측정으로 시행되어왔다. 차세대염기서열분석법(massive parallel sequencing, MPS)이 도입되어 더욱 많은 STR 좌(loci)를 분석할 수 있게 되고, 절편길이 다형성 외에도 변이를 분석하는 것도 가능해지게 되었다. MPS의 이점과 점점 낮아지는 비용으로, 법의학 분야에서 MPS를 이용한 접근이 활발해지고 있다. Thermo Fisher Scientific Inc.에서 조상찾기패널을 포함한 법의학용 단일염기다형성(single nucleotide polymorphisms, SNP) 상업 키트를 출시하여 법의학 분야에서 이미 널리 이용되고 있으며, 고해상도 분석을 위해 커스텀 MPS-SNP 패널로 개인식별검사를 시행하기도 한다.
31 상염색체 STR (autosomal STRs, A-STRs)과 4 성결정 표지자로 구성된 Precision ID GlobalFiler™ MPS STR Panel v2 (Thermo Fisher Scientific, Waltham, USA)와 22 A-STRs, 23 Y-STRs, 그리고 amelogenin으로 구성된 PowerSeq™ Auto/Y system (Promega, Madison, USA)이 잘 알려진 MPS-STR 패널이다. 혈연관계 분석에 쓰이는 ForenSeq™ DNA Signature Prep Kit(Illumina, San Diego, USA)은 A-STRs, X-STRs, Y-STRs 외에도 SNPs(identity-informative SNPs 및 phenotypic-informative SNPs)이 결합된 형태의 시약으로, 널리 평가되고 있다. 추가 분석 좌위가 많아질수록 개인식별검사의 증거력이 높아지기 때문에 STRs에 SNPs를 혼합한 형태의 MPS 패널의 전망이 밝으며 이에 저자들은 in-house로 이러한 MPS 혼합 패널을 개발해보았다.
우선 패널을 디자인하기 위해 Expanded CODIS (Combined DNA Index System) core STR loci 전부 및 중국 법의학회에서 권장하는 A-STRs를 1차적으로 선정하고 법의학적으로 중요한 좌위들을 추가하였다. X 및 Y STR 좌위 선정에 있어서는 일반적으로 사용하는 25개 Y-STRs과 7개 X-STRs를 포함시켰다. SNP 표지자 는 저자들의 이전 연구에서 사용한 273 SNPs와 HapMap (http://hapmap.ncbi.nlm.nih.gov/), dbSNP (http://www.ncbi.nlm.nih.gov/snp/) 등을 참고하여 296 SNPs (239 A-SNPs, 30 X-SNPs 27 Y-SNPs)를 선정하였다. Illumina사의 Design Studio (https://designstudio.illumina.com/)와 TruSeq Custom Amplicon을 이용하여 프라이머와 라이브러리를 디자인하였다.
검체는 혈연관계가 없는 한족 50명(남 24, 여 26) 의 말초혈액을 사용하였으며, 컨트롤로 Promega 사의 Human genomic DNA (gDNA) 9947A와 9948 사용하였다. 기존 검사법과의 비교를 위해 SiFaSTR™ 23-plex system (Academy of Forensic Sciences, Ministry of Justice, P.R. China), Goldeneye™ DNA ID 22NC Kit (Goldeneye Co. Ltd, China), AGCU 21 + 1 STR Kit (AGCU ScienTech Co. Ltd, China), Investigator Argus X-12 kit (Qiagen) 및 AmpFlSTR Yfiler Plus PCR Amplification Kit (Thermo Fisher Scientific) 검사를 병행하였다. SNP calling 검증은 이전 연구결과와 Sanger 시퀀싱으로 시행하였다.
민감도 평가를 위해 gDNA 9948을 희석하여 10ng, 5 ng, 2 ng, 1 ng, 500 pg, 200 pg,로 준비하여 각각 라이브러리를 제작, 염기서열 분석을 시행하였다.
검체 간의 수행능 차이는 손톱, 모발, 혈흔 검체 및 친자확인용 trio 검체로 평가하였다. 혼합 검체 평가를 위해 gDNA 9947A (여성)과 9948 (남성)의 검체를 1:1, 1:3, 3:1, 1:9, 9:1, 1:19, and 19:1로 혼합하여 평가하였다.
Illumina 사의 TruSeq Custom Amplicon Low Input Kit을 사용하여 DNA 라이브러리를 제작하였고 Agilent Bioanalyzer 2100 (Agilent Technologies, CA, USA)로 절편 사이즈를 분석하였다.
얻어진 데이터는 커스텀한 “STRsearch” 파이프라인으로 분석하였는데 그 과정을 간단히 설명하자면 다음과 같다: 1) GRCh37을 레퍼런스로 매핑; 2) STRallocation; 3) STR read 병합; 4) STR read 검색(디폴트 100x); 5) 필터(디폴트 < 1%); 6) variant calling; 7) 보고서 작성. SNPs은 또 다른 자체개발 python 파이프라인으로 분석하였으며 그 과정은 다음과 같다: 1) BWA-MEM (http://bio-bwa.sourceforge.net/)을 사용하여 GRCh37을 레퍼런스로 매핑; 2) 정렬된 리드를 추출; 3) GATK 4.0 (https://software.broadinstitute.org/gatk/)을 사용하여 SNP 유전형 calling을 실시. 이형접합체 평형(heterozygote balance, Hb)은 STR은 더 많은 allele에 대한 소수 allele의 비로 나타내었으며, SNP은 A, C, G, T 순으로 나타내었다. 본 연구에서 stutter는 n-1만 분석하였다. R software v 3.6.2 (https://www.r-project.org) 혹은 Python v 3.7.4 (https://www.python.org)로 readsRelevant plot 만들어 noise read하였고 그 외 HWE, LD, PD, PEduo, PEtrio 등도 평가하였다. 프라이머와 증폭산물에 관한 자세한 내용은 원 논문의 표를 참고할 수 있다1.
연구 시작시점에서 94 STRs과 296 SNPs을 계획하였으나, NT 반복수가 높다든가 인근 GC 함량의 문제로 최종적으로 SiFaMPS 패널 프로토타입은 87 STRs, 294 SNPs, amelogenin으로 SifaMPS 패널을 구성하였다.
염기서열분석 수행능과 결과 평가 디폴트 세팅 DoC ≥ 100 reads, stutter ratio < 0.5, noise < 1%로 STR 분석을 수행하였더니 81 (2.13%) 좌위에서 dropout이 발생하였다. 가시적인 개요는 아래 그림과 같다.
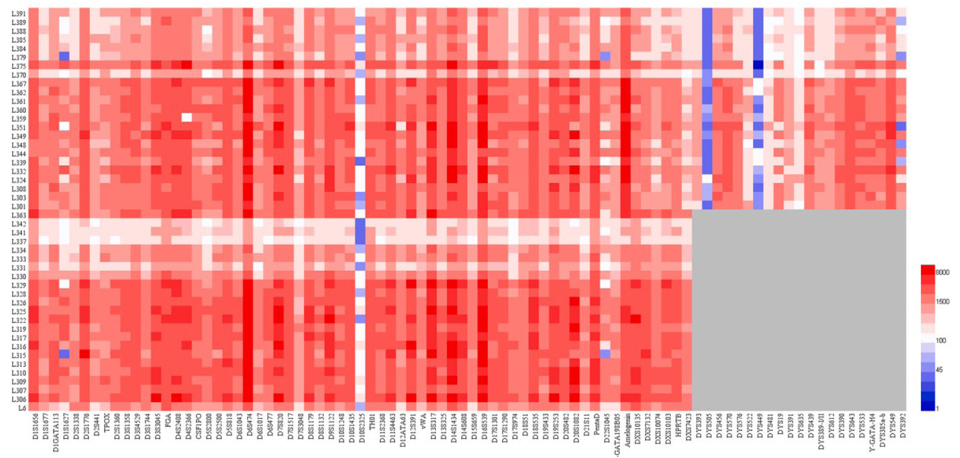
최소 DoC를 45 reads로 수정해도 여전히 38개의 dropout이 관찰되어 해당 좌위는 분석에서 제외하였다. SNP calling에서는 100 reads 이상의 coverage, 0.3-3의 Hb threshold, 0.9 이상의 FMAR를 적용하였더니 5.45%가 dropout되고 나머지 결과는 일치하였다. 다음 그림에서 SNP calling 의 전체적인 수행능과 Chr7의rs730437, Chr8의 rs4288409, Chr10의 rs7088884의 read가 나머지에 비해 낮은 것을 볼 수 있다.

DoCs는 평균적으로 58 A-STRs, 6 X-STRs, 23 Y-STRs로 계산되었다. 좌위에 따라 DoC 불균형이 심했는데 dropout 발생율이 높고 DoC가 매우 낮은 좌위들을 배제하였다. 좌위간 불균형이 여전히 존재하긴 했지만, 85 STRs에 대한 유전형 분석에 큰 문제는 없었다.
게다가, 아래 그림에서 보다시피 85 STRs에 대한 SCRs 평가를 시행하였는데 모든 STRs에서 60% 이상의 대립유전자비(allele ratio)를 보였다.
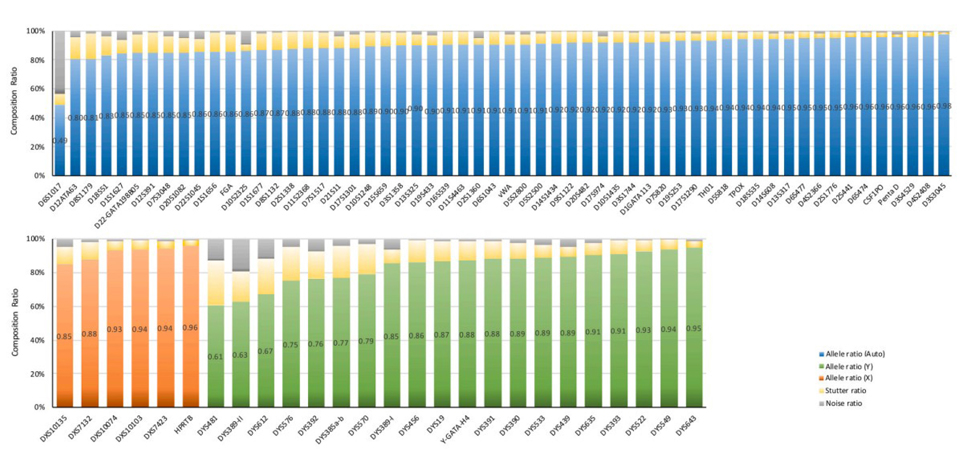
STRinNGS v2.0로 D6S1017 좌위를 분석한 바, 비슷하게 낮은 대립유전자비(<50%)가 관찰되었는데, D6S1017 downstream 부위에 무작위 변이가 발생하는 것으로 보아 참대립유전자 검출을 하지 못해 발생한 오류로 생각된다. “n-1” stutter 분석에서 삼염기반복 모티프의 stutter reads 발생율이 높았으며, 각각 A-STRs의 7.65%, X-STRs의 6.55%, Y-STRs의 11.71%이었다.
다음 그림에서 58 A-STRs과 6 X-STRs 좌위에 대한 Hb를 볼 수 있다.

Hb의 중앙값은 0.54-0.92였고, Hb 값이 낮은 D10S2325가 가장 낮은 평균 coverage를 보였는데, 이는 기존 연구 결과와 일치했다. 64 STRs의 Hb 중앙값 중 0.6 미만은 단 3건으로 전체적으로 좋은 수행능을 보여주었다. A-SNPs와 X-SNPs는 Hb의 변이가 컸는데(0.11-6.38), reads가 45 이상이면 모두 분석에 포함시켜, 아웃라이어(Hb < 0.3 혹은 > 3)를 배제하지 않았기 때문이다. 불균형이 심한 좌위를 배제한 경우, filtering을 시행하지 않아도 SNP 유전형분석에 안정적인 Hb 값을 획득할 수 있었다.
50명의 비혈연관계 한족의 검체에서 PCR-CE와 SifaMPS 패널 프로토타입으로 구한 유전형을 비교하였을 때, 81.69% (5522/6760)의 대립유전자에서 결과가 일치하였다. STRs 85 좌위 중 69 좌위에서 결과가 완전히 일치하였고, 18.31%의 불일치 결과는 세 가지로 나뉜다: 1) 절대다수(96.93%)는 두 방법 간의 명명법 차이(12 개의 non-CODIS 좌위에서 발생)로 인한 것이었고; 2) STR이나 그 인접부위에 변이가 있는 경우가 있었으며; 3) 대립유전자 dropout이 발생한 경우가 한 건 있었다. 흔하게 관찰되는 다수의 NT 결실의 경우, 대립유전자 길이가 짧아지므로 분석 시 주의해야 한다. 특히 non-CODIS STR 좌위를 사용하여 MPS로 분석하는 경우, 서열 병합과 CE와의 비교를 극도로 주의하여 시행해야 한다. SNP 유전형 비교는 100%에서 일치하였다.
9947A와 9948 gDNA를 반복분석하여 재현성을 검증한 바, STR 결과는 완전히 일치하였다. SNP를 반복분석한 결과 비교에서도, 모든 유전형에서 결과가 일치하였다.
PCR 조건을 29 cycles로 고정하고, 콘트롤 DNA9948 분주량을 다르게 하여 패널의 민감도를 평가하였고 10 ng과 5ng DNA를 분주한 경우 완전한 STR 프로 파일을 얻을 수 있었다. 2 ng에서 200 pg 사이의 분주량에서는 74.1-98.9%의 STR이 분석기준을 넘겨 정확한 분석이 가능했다. 예상한 바와 같이, DNA 분주량이 줄어들수록 Hb 변동이 심해지고 1ng 이하의 DNA 분주 시 대립유전자 불균형이 심하게 관찰되었다. SNP 분석에서도 분주량이 적어질수록 Hb 변동이 심해졌으며, 500 pg 이하의 분주량부터 불일치가 발생하여 500 pg 분주 시 2.4%. 200 pg 분주 시 5.5%의 SNP 유전자형 불일치가 있었으며 그 이상의 분주량에서는 유전형이 모두 일치하였다. SNP 분석 시, calling 조건을 넓힐수록 더 많은 유전형이 분석에 포함되어, 부정확한 결과 또한 늘어났다. 이는 특히 법의학 검사에서 권장하지 않기 때문에, 결과의 정확성을 보장하기 위하여 저자들은 >45 reads, 0.3 ≤ Hb ≤ 3의 calling 조건을 채택하였다. 전체적으로 85 STRs와 293 SNPs에 대한 ull-sequence 프로파일을 얻기 위해서는 최소 10 ng 이상의 DNA가 필요했다. 이러한 결과는 1 ng이상의 DNA를 요구하는 기존의 ForenSeqTM DNA Signature Prep Kit 만큼 SifaMPS 프로토타입 패널의 민감도가 좋지는 않다는 것을 것을 나타내며, DNA 추출 방법의 차이에 기인한 것으로 생각된다. 민감도는 친자검사보다 법의학적 검사에서 문제가 될 수 있는데 이를 해결하기 위해 다음과 같은 조치를 취해볼 수 있다: 1) PCR 사이클 횟수를 올려보기; 2) 유전형 calling criteria 를 완화하기.
여러 명의 검체가 혼합된 것으로 시행하는 검사는 범죄현장에서 발견된 증거에서 특히 중요한데, 9947A와 9948 DNA를 1:1 부터 1:19 까지 혼합하여 검사해본 바 D10S2325를 제외한 부위에서 STR은 모두 감별해낼 수 있었다. 남성의 DNA 양이 적은 혼합물의 경우 감별력이 조금 낮아졌는데, 그 이유는 패널에 포함된 Y-STR 좌위 개수가 A-STR이나 X-STR보다 적기 때문인 것으로 생각된다. SNP 감별에서는 Hb와 FMAR 척도가 검체가 혼합되었음을 알 수 있는 중요한 지표였다. 검체 유형에 의한 패널 수행능 또한 법의학에 중요한 내용으로, 모발, 손톱, 혈흔 검체를 비교해 본 결과, 손톱 검체의 결과는 좋지 않아 대상 검체에서 제외되었고, 모발과 혈흔에서는 일관된 결과를 얻을 수 있었다.
본 검사로 저자들은 MiSeq FGx System에 기반한 새로운 MPS 프로토타입 패널을 개발하였고 이는 기존 상업키트보다 더 많은 유전정보를 제공하여, 법의학 분야의 개인식별 검사와 친자검사에 도움이 될 것으로 생각된다. 다만, 12개의 non-CODIS STRs 에서 최신 법의학 STR 서열구조 가이드(https://strider.online)와 다른 명명법을 사용했는데, 해당 좌위들이 현재 사용 중인 상업키트에도 포함되어 있는 만큼 MPS와 PCR-CE검사 결과 비교 및 합치를 위해서 결과 기술에 주의를 기울여야 하겠다.
[References]
1. Tao R et al. Parallel sequencing of 87 STR and 294 SNP markers using the prototype of the SifaMPS panel on the MiSeq FGx™ system. Forensic Sci Int Genet. 2021 May;52:102490.
