Highly Sensitive Methylation Detection Using Enzymatic Methyl-seq and Twist Target Enrichment
Introduction DNA methylation은 유전자 발현을 조절함으로써 세포성장에 있어서 중요한 역할을 담당한다. 어떤 경우에는, cytosine methylation의 오류로 인해 발암성 유전자가 발현될 수도 있다. 하지만 환자 샘플에서 이러한 잘못된 변화를 확인하는 것은 어려웠었다. 차세대 염기서열 분석법(NGS)이 sodium bisulfite가 처리된 DNA 상의 methylated cytosine을 검출하는데 효과적인 수단임이 입증되기는 하였지만, 이 화학적 변환과정은 DNA 손상을 야기하여 GC bias를 불러오고 complexity도 떨어지기 때문에 민감도가 제한적이 된다[1].
트위스트사(Twist Bioscience사)의 targeted methylation workflow에서는 liquid biopsy를 통한 암 검사와 같은 새로운 솔루션을 포함하여 다양한 환경에서 메틸롬(methylome) 분석을 위해 high complexity와 uniformity를 보이는 라이브러리를 생산하는 전체 솔루션을 소개한다. 이 전체 프로토콜은 enzymatic conversion방법을 Twist custom methylation panel을 이용한 hybrid-capture과정에 적용한 것이다. 이를 기존의 bisulfite conversion방법과 비교하여 설명하고 이 공정이 갖는 장점을 sequencing metrics를 통해 확인하였다
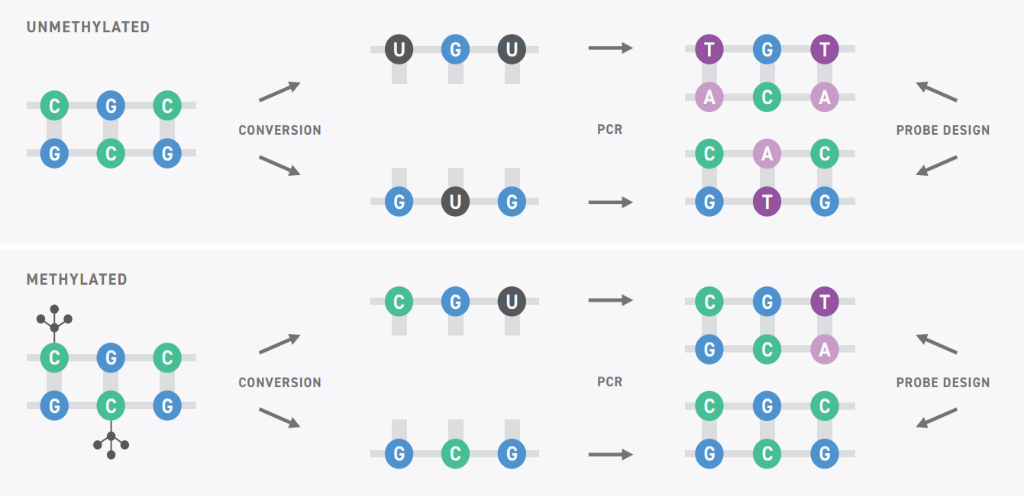 그림1. Methylation Conversion. Methylation sequencing은 enzymatic 또는 chemical방법으로 unmethylated cytosine을 deamination 시켜 uracil로 변환시키는 반면 methylated cytosine은 그대로 유지시킨다. 증폭 단계에서 uracil은 complementary strand의 adenine과 결합하여 unmethylated cytosine이 thymine으로 치환된다. 변환 이후의 최종 산물은 비대칭형으로서 두 개의 서로 다른 double stranded DNA를 만든다(윗줄).
그림1. Methylation Conversion. Methylation sequencing은 enzymatic 또는 chemical방법으로 unmethylated cytosine을 deamination 시켜 uracil로 변환시키는 반면 methylated cytosine은 그대로 유지시킨다. 증폭 단계에서 uracil은 complementary strand의 adenine과 결합하여 unmethylated cytosine이 thymine으로 치환된다. 변환 이후의 최종 산물은 비대칭형으로서 두 개의 서로 다른 double stranded DNA를 만든다(윗줄).
methylated DNA는 동일한 과정을 거쳐 추가 염기서열이 생성된다 (아랫줄)
Result
Library Preparation for Methylation Detection
전통적 프로토콜에서는 bisulfite를 처리해 화학적으로 unmethylated cytosine을 uracil로 변환함으로써 methylated cytosine residue를 확인한다. PCR 후, unmethylated cytosine은 thymine으로 염기서열이 읽혀지고, methylated cytosine은 cytosine으로 읽혀진다 (그림 1)
그러나, bisulfite처리는 종종 sample preparation을 어렵게 만들 수 있는 DNA break를 야기하여 결국 methylation 검출까지도 어렵게 한다[2]. Twist Bioscience사는 New England Biolabs사와 협력하여 NEBNext® Enzymatic Methyl-seq (EM- seqTM)을 출시하였다. Sample 손상을 야기하는 chemical conversion 과정 없이도, bisulfite 처리와 동일한 변환 결과를 가져와 훨씬 좋은 최종 결과를 얻게 해준다.
methylation detection을 위한 라이브러리 제작은 다음의 6가지 주요 단계로 구성된다.

Enzymatic method와 bisulfite method은 비슷한 효율로 unmethylated cytosine을 thymine으로 변환시킨다(그림 2). 그러나, enzymatic method로 변환된 library는 GC비율이 높은 부위에서 증가하고 더 균일한 커버리지를 보여준다(그림 3). 이것은 잠재적으로 이러한 부위에서 DNA break가 덜 일어난 결과일 것이다.
Twist Methylation system은 라이브러리 제작과 enzymatic conversion 전에 mechanical shearing을 하기 때문에 fragment size가 다양하게 생겨난다. methylation detection을 위해 이 라이브러리 제작 방법을 사용할 때, 실제 fragment size는 초기 DNA가 얼마나 온전한 상태인가에 따라 결정되지만, Twist Bioscience사는 약 200 ~300bp 크기로 DNA를 fragmentation해서 최종 DNA fragment가 350bp ~ 450bp정도가 되는 것을 권장한다. 라이브러리 수율은 초기 DNA의 양과 라이브러리 제작 중에 사용하는 PCR cycle수에 따라 달라진다. Twist는 DNA농도, 200ng, PCR cycle 수를 9회로 시작하기를 권장한다. 이 권장사항은 다음 hybrid capture단계에서 capture할 수 있는 충분한 양의 DNA가 존재할 수 있도록 하기 위함이다. 최종 QC 단계에서는 fragment size를 (평균 peak size, 375bp) 확인한다.
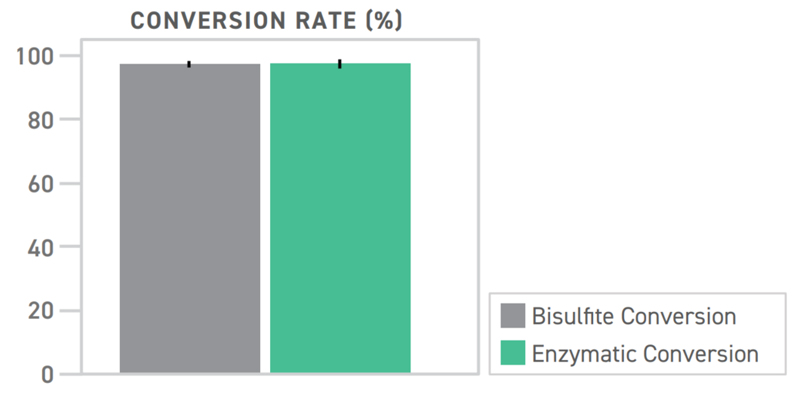 그림2. Conversion Rate Comparison for Bisulfite & Enzymatic Conversion. 두가지의 library 변환 방법 모두 non-CpG site에서 cytosine이 thymine으로 99.9% 이상 변환되었다.
그림2. Conversion Rate Comparison for Bisulfite & Enzymatic Conversion. 두가지의 library 변환 방법 모두 non-CpG site에서 cytosine이 thymine으로 99.9% 이상 변환되었다.
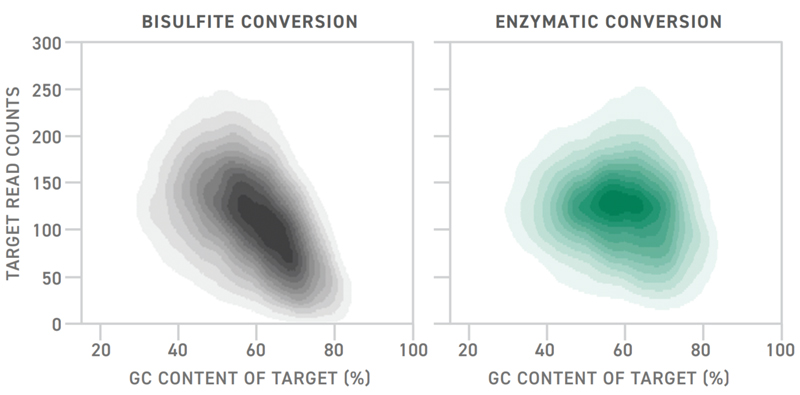 그림3. Coverage by Target GC Content for Bisulfite and Enzymatic Conversion. 두가지의 library 변환 방법은 모두 Twist Fast Hybridization target enrichment 적용가능하다. 그러나 enzymatic conversion방법을 사용해 제작한 library에서 hybrid selection metrics의 개선이 관찰된다. Bisulfite conversion method(gray)를 사용할 때는 high GC target region에서 coverage가 낮고, enzymatic conversion method(green)을 사용했을 때 bias가 낮게 나타난다.
그림3. Coverage by Target GC Content for Bisulfite and Enzymatic Conversion. 두가지의 library 변환 방법은 모두 Twist Fast Hybridization target enrichment 적용가능하다. 그러나 enzymatic conversion방법을 사용해 제작한 library에서 hybrid selection metrics의 개선이 관찰된다. Bisulfite conversion method(gray)를 사용할 때는 high GC target region에서 coverage가 낮고, enzymatic conversion method(green)을 사용했을 때 bias가 낮게 나타난다.
Use of the Control DNAs in the Twist Targeted Methylation Sequencing Workflow 불완전 conversion이 되면, 변환되지 않은 unmethylated cytosine 은 methylation된 것으로 잘못 해석되기 때문에 분석에 있어서 false positive rate을 증가시킨다. 이를 줄이기 위해 methylation level이 알려진 standard DNA를 사용하여 conversion rate을 확인할 수 있다. Twist Targeted methylation sequencing workflow 의 일부과정으로서, NEB EM-seq methylation library preparation kit는 CpG methylated pUC19 DNA와 unmethylated Lamda DNA를 optional control을 포함하고 있다. 이 두개의 control 모두 methylation level이 알려져 있기 때문에 sequencing 이후에 이들을 이용하여 conversion rate을 정확하게 판단할 수 있다. Target enrichment panel에 complementary probe가 부족할 수도 있기 때문에, 이 control 들은 hybrid capture에는 사용하지는 않아야 한다. 대신, hybrid capture 후에, sequencing을 위한 sample과 함께 섞는다.
Twist Workflow에서, 이 control의 유용성을 보여주기 위해, Twist NEBNext Enzymatic Methylseq Library Preparation Protocol의 appendix A 를 참고하여 라이브러리를 제작하였다. 각 control DNA를 48㎕씩 한 튜브에 섞은 후 vacuum concentrator를 이용하여 건조시켰다. 건조시킨 DNA를 50 ㎕의 0.1X TE (pH 8.0) buffer에 녹인 후 라이브러리 제작과정을 진행하였다.
표 1은 measured conversion efficiency vs.expected conversion efficiency과 sequencing 후의 methylation level을 보여준다. EM-seq은 두 control에 대하여 99.5%를 넘는 conversion rate 로 예상한 효율을 나타냈다. Unmethylated Lamda DNA와 CpG methylated pUC19 DNA control의 expected CpG methylation level은 각각 0.5%와 95~98%였다. 두 control의 CpG methylation level의 측정수준과 기대수준은 일치하였다. 분석은 methylation caller를 이용하여 계산하였다. 이는 알려진 methylation level의 control DNA는 1)conversion 과정이 완벽하고, 2)분석에 있어서 false positive를 최소화된 것을 확인하는데 사용할 수 있음을 보여준다. Twist는 EM-seq process 중에 enzymatic conversion rate에 대하여 확신이 없는 경우에는 이 control을 사용하도록 권장한다.
| METRIC | UNMETHYLATED LAMBDA DNA | CpG METHYLATED pUC19 DNA |
|---|---|---|
| Expected Conversion Effciency | >=99.5% | >=99.5% |
| Measured Conversion Effciency | 99.77% | 99.57% |
| Expected CpG Methylation Level | Up to 0.5% | 95-98% |
| Measured CpG Methylation Level | 0.22228% | 95.7572% |
표 1. Conversion 효율 및 CpG methylation 의 기대치와 측정치 비교
Twist Target Enrichment for Methylation Detection Twist Targeted Methylation Sequencing Workflow는 고감도의 customized methylation detection을 위해 Twist Fast hybridization Target Enrichment System을 이용한다. 이 시스템은 이전의 workflow보다 더 빠른 hybridization 시간에도 불구하고(이전보다 감소된 hyb시간) sequencing metrics에 영향을 주지 않고 동등 이상의 결과를 확인할 수 있다.
Twist Fast Hybridization Target Enrichment System은 다음의 5단계로 이뤄져 있다.
이 시스템은 최대 8개의 sample을 pooling해 실험을 진행할 수 있고, manual시간과 pipetting횟수도 감소시켰다. Twist Targeted Methylation Sequencing protocol은 다양한 맞춤형 메틸화 패널에 적합한 최고의 관리기준을 적용하기를 권장한다. 그러나, 모든 target capture system에서와 마찬가지로, target size 및 methylation level을 포함한 다양한 요인들이 최종 capture data metrics에 영향을 미칠 수 있다. 이러한 요인들이 시퀀싱 효율에 미치는 영향은 아래에서 논의한다.
Target Region Size custom target region과 관련된 많은 요인들이 최종적인 targeted sequencing metrics에 영향을 미치기 때문에, 가장 좋은 결과를 얻기 위해서는 최적화가 필요할 수 있다. 예를 들면, 매우 작은 패널(<0.5 Mb) 또는 target region 상에 high GC content을 가진 패널은 패널 설계 상의 작은 변화에도 특히 민감하다. 포괄성과 off-target control 사이에서 어떻게 최적의 균형을 이루는 가는 target 영역의 특징과 패널의 적용 목적에 달려있다. 예를 들어, 패널 설계 과정에서 중간크기의 패널과 적은 수의 샘플을 다루는 연구자는 늘어난 off-target capture의 비율을 맞추기 위해 추가 시퀀싱이 필요할 수 있음에도 불구하고 패널에 대해 일정한 probe수를 유지하는 것을 선호할 수 있다. 반면, 훨씬 작은 패널(이 경우 off-target capture가 다른 패널에 비해 필요한 시퀀싱양이 더 빠르게 증가)을 다루는 연구자들이나, 아주 많은 수의 샘플(이 경우 비용이 증가 할 수 있음)을 다루는 연구자들은 비용을 최적화하기 위해 설계조건을 더 엄격하게 하는 것을 선호할 수 있다.
패널 크기와 sequencing metrics 간의 관련성을 평가하기 위해, 3개의 다른 패널을 Twist Targeted Methylation Sequencing Workflow과 함께 사용하였다. 각 패널들은 다양한 크기의 methylated target 영역을 포함한다: 0.5Mb, 3Mb, 50Mb (그림 4). 이 연구에서 사용된 가장 큰 패널은 off-target level이 7% 정도였고, 전체 패널의 off-target level은 10% 미만으로 확인하였다. 모든 target size에 대해 Capture uniformity(fold-80 base penalty)은 그 값이 1.4~1.7으로 우수하였다. 30X coverage 비율은 모든 패널에서 90%를 넘었다. 그러나, 패널 크기에 따른 duplication rate의 증가는 어떤 key metrics를 우선적으로 다뤄야 할지를 결정하는 데 있어서 중요한 고려사항이다.
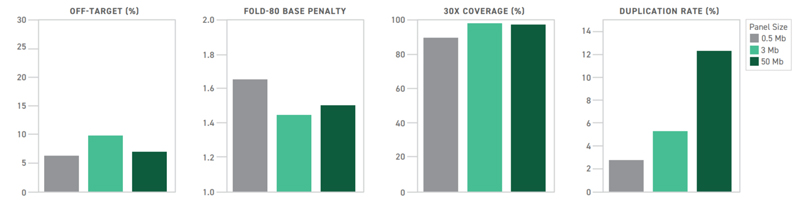 그림4. Size of Target Region within Custom Panel and its relationship to Picard Metrics. Twist Targeted Methylation Sequencing Protocol에 따라 및 single-plex reaction으로 실험을 진행하였다. 0.5Mb, 3Mb, 50Mb의 methylation panel에 대한 capture metrics.
그림4. Size of Target Region within Custom Panel and its relationship to Picard Metrics. Twist Targeted Methylation Sequencing Protocol에 따라 및 single-plex reaction으로 실험을 진행하였다. 0.5Mb, 3Mb, 50Mb의 methylation panel에 대한 capture metrics.
Identification of differing Target Region Methylation Level Methylation level genome에 따라 다양하다. Methylation level 차이를 이용하여 특정 암을 조기에 발견할 수 있기 때문에, methylation detection에 사용하는 프로토콜이 custom panel 디자인에 적합해야 하고, hyper와 hypo-methylated region을 확인할 수 있어야 한다는 점은 필수사항이다. unmethylated region의 변환은 sequence complexity를 감소시키고 hybrid capture단계에서 downstream문제를 야기할 수 있다. 그러나, 이 문제는 라이브러리 제작 시약, hybrid capture 시약, custom panel 디자인을 적절하게 조화시킴으로써 줄일 수 있고, 이를 통해 디자인된 panel의 probe가 다양한 범위의 AT/GC rate와 methylation level을 가진 부위 전체를 고르게 분포(capture)할 수 있게 할 수 있다. Twist사의 targeted methylation custom panel은 target 에 대해 4개의 probe가 디자인되어 있어서 conversion 이후, sense와 anti-sense DNA에 대한 methylated-, unmethylated 상태의 DNA를 모두 capture할 수 있다 (그림 1, 우측 참조).
Twist Targeted Methylation Sequencing Workflow을 통해, 균등한(evenness) coverage를 확인하였다. 이를 위해Enzyme와 bisulfite 처리 방식의 2가지 conversion system을 이용하여 genomic DNA를 conversion하였고, 1.5Mb custom panel로 hybrid capture 실험을 진행하였다. hyper-, hypo-methylation 영역에 대한 target coverage를 확인하였다.
그림 5는 EM-seq conversion 방법을 사용했을 때, target read counts가 hyper- 와 hypomethylation region 모두에 대해 균등하게 분포되어 있음을 보여준다(녹색). 반면, 많이 사용되고 있는 bisulfite conversion방법(회색)의 결과에서는 target read counts가 상대적으로 균등하지 않았다. 따라서, Twist custom panel 디자인은 Twist Targeted Methylation Sequencing Workflow과 함께 사용했을 때, target methylation level에 관계없이 전체 target DNA들을 효과적으로 capture 한다.
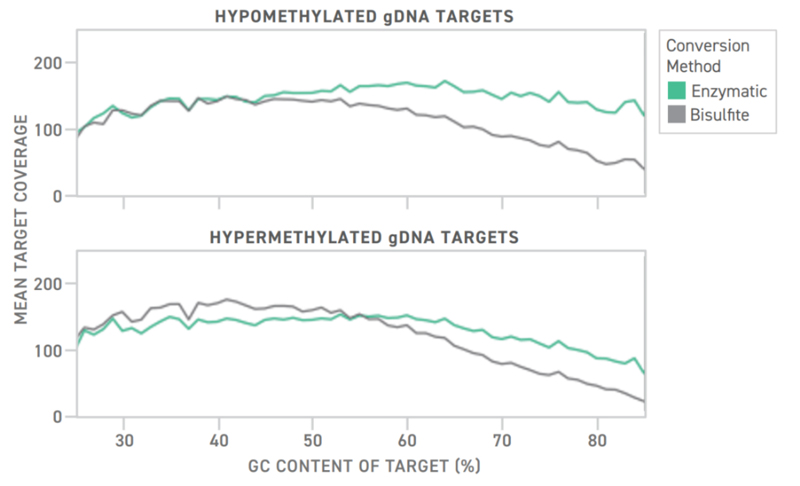 그림5. Coverage by Target GC Content for Hypo- and Hypermethylated gDNA Libraries Prepared with Enzymatic and Bisulfite Conversion Techniques. Twist사의 enzymatic 라이브러리 제작 방식을 사용하였을 때, hypo-, 와 hyper-methylated gDNA 모두 1.5Mb에 대한 target coverage 가 모든 GC rate에 걸쳐 더욱 고르게 분포되어 있다.
그림5. Coverage by Target GC Content for Hypo- and Hypermethylated gDNA Libraries Prepared with Enzymatic and Bisulfite Conversion Techniques. Twist사의 enzymatic 라이브러리 제작 방식을 사용하였을 때, hypo-, 와 hyper-methylated gDNA 모두 1.5Mb에 대한 target coverage 가 모든 GC rate에 걸쳐 더욱 고르게 분포되어 있다.
Twist Targeted Methylation Sequencing Workflow를 다양한 methylation level을 대상으로 한 결과를 확인하기 위해, 다양한 methylation level을 가진 라이브러리를 제작한 후 1Mb 패널을 이용하여 capture하였다. 이들 라이브러리는 hypo- and hypermethylated control human cell lines(EpiScope® Unmethylated HCT116 DKO gDNA PN# 3521 and EpiScope Methylated HCT116 gDNA PN# 3522, Takara Bio USA)을 혼합하여 제작하였고, 이를 통해 0, 25, 50, 75, 100% methylation level을 포함하는 라이브러리를 만들었다. Key capture metrics를 나타낸 그림6은 methylation level과 최종 Picard metrics 사이에 관계가 거의 없음을 보여준다. 이 데이터는 Twist Methylation System이 hypo- and hypermethylated DNA regions 에 모두 사용될 수 있으며, 정확한 결과를 확인할 수 있음을 보여준다.
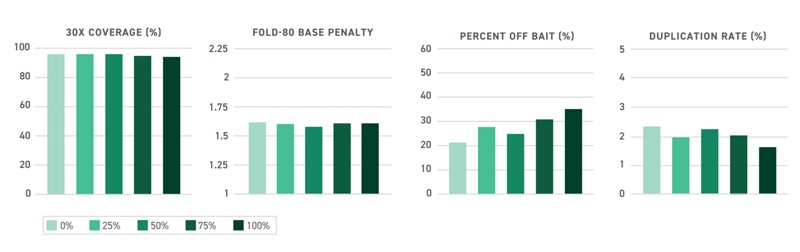 그림6. Picard HsMetrics for gDNA Libraries with Variable Methylation Levels. EM-seq conversion 방법으로 0, 25, 50, 75, 100% methylation rate을 가진 hypo-/hypermethylated cell lines을 혼합하여 라이브러리를 제작하였고, Twist target enrichment library prep 방법에 따라 1MB custom panel을 사용해 capture 하였다. CpG methylation level이 서로 다르지만, 라이브러리 유형에 따라 key hybrid selection metrics는 일정하다.
그림6. Picard HsMetrics for gDNA Libraries with Variable Methylation Levels. EM-seq conversion 방법으로 0, 25, 50, 75, 100% methylation rate을 가진 hypo-/hypermethylated cell lines을 혼합하여 라이브러리를 제작하였고, Twist target enrichment library prep 방법에 따라 1MB custom panel을 사용해 capture 하였다. CpG methylation level이 서로 다르지만, 라이브러리 유형에 따라 key hybrid selection metrics는 일정하다.
Conclusion Twist Bioscience사는 NEB사의 enzymatic conversion과 custom methylation panel 및 최적화된 target enrichment 시약을 포함하는, 모든 과정의 시약과 panel로 구성된 All-in-One kit를 제공하고 있다.
Twist Targeted Methylation Sequencing Workflow은 highly sensitive methylation detection이 가능하고, 이는 시간적 제약을 극복하기 위해서나 hands-on time을 줄이기 위해 필요에 따라 변형할 수 있다. 이 공정을 이용하면, custom panel 디자인의 target size와 methylation level이 최종 Picard metrics에 거의 영향을 미치지 않는다. 그러므로, 이 시스템은 methylation detection에 관심이 있는 다양한 연구자들이 도입해, 연구에 활용하기에 매우 적절하다.
# Reference
1. Vaisvila R, Pommaluri V K C, Sun Z, Langhorst B W, Saleh L, et al. (2019). EM-seq: Detection of DNA Methylation at Single Base Resolution from Picograms of DNA. BioRxiv. https://www.biorxiv.org/content/10.1101/2019.12.20.88469 2v1.full.pdf (accessed 10th Feb 2021).
2. Tanaka K & Okamoto A (2007). Degradation of DNA by bisulfite treatment. Bioorganic & Medicinal Chemistry Letters, 17(7), pages 1912–1915.
