연세의대 세브란스병원 진단검사의학과
Long-read sequencing 검사의 정의 및 특성 Long-read sequencing (LRS) 검사는 기존 short-read next-generation sequencing (SR-NGS) 검사의 짧은 분석 범위 단점을 보완한 것으로서 긴 DNA 단일 분자 가닥의 염기서열을 한번에 실시간으로 읽을 수 있는 기술이다. 2세대 염기서열 분석법으로도 알려진 SR-NGS는 75~400 염기쌍의 길이를 가진 조각들을 동시 대량으로 분석하는 방법으로서 저렴한 비용으로 99.9% 이상의 높은 정확도로 염기서열을 읽을 수 있기 때문에 주로 작은 크기의 유전자 변이 (single-nucleotide variants, small indels, copy number variants)를 검출하는데 이용되어 왔다 [1]. 반면 3세대 염기서열 분석법으로 알려진 LRS는 10,000 염기쌍 이상의 길이를 가진 조각들의 염기서열을 PCR 증폭없이 그대로 읽을 수 있다. 따라서 새로운 유전체 조립 (de novo assembly), 홑배 수체형 위상 (haplotype phasing) 확인, 구조적 변이 (structural variant) 확인, 일렬반복 부위 확장 (tandem repeat expansion) 확인, 유사유전자 (pseudogene) 구분 등에 유리하다 [2]. 하지만 LRS는 일반적으로 SR- NGS에 비하여 개별 염기서열의 낮은 정확도와 비싼 비용 때문에 아직은 연구용으로 더 많이 사용되고 있다.
Long-read sequencing 검사의 종류 LRS는 Pacific Biosicences사의 PacBio SMRT sequencing과 Oxford Nanopore Technologies사의 Nanopore sequencing이 대표적이다. 이들은 각기 다른 방법으로 염기서열을 분석하기 때문에 한번에 읽는 염기의 수, 분석의 정확도 등에 차이가 있다.
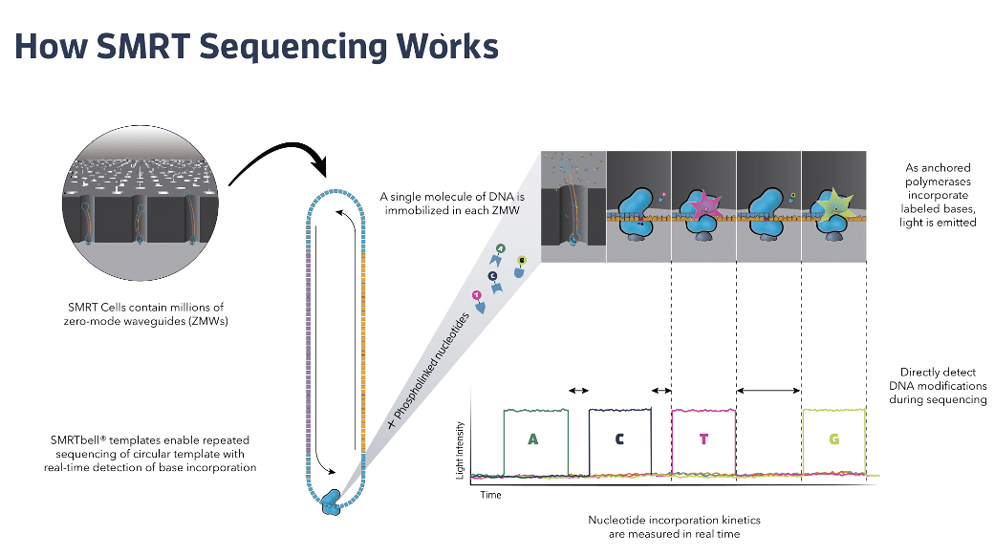 그림1. Pacific Bioscience 장비의 원리 (Pacific Biosicences사의 허가를 받아 사용)
그림1. Pacific Bioscience 장비의 원리 (Pacific Biosicences사의 허가를 받아 사용)
Pacific Bioscience PacBio SMRT sequencing은 single-stranded hairpin 구조의 어뎁터가 양쪽 끝에 위치하고 분석하려는 double-stranded DNA가 삽입된 원형의 DNA molecule template (SMRTbell)을 이용한다 [3]. PabBio 장비에는 zero mode waveguides (ZMW)라고 불리는 무수히 많은 웰 (well)이 있는데 바닥에 DNA 중합효소가 부착되어 있다. SMRTbell은 이 중합효소에 결합되고 다른 형광이 부착된 dNTP (dATP, dCTP, dTTP, dGTP) 염기가 SMRTbell에 상보적으로 삽입된다. PacBio 장비는 dNTP가 삽입되면서 발생하는 형광을 검출하여 실시간으로 염기서열을 판독한다. SMRT sequencing은 최근에 에러를 감소시키기 위해 삽입된 DNA를 여러번 반복해서 읽어내는 circular consensus sequencing (CCS) 방법을 도입하여 정확도를 99% 이상으로 향상시키기도 했다 [4].
Oxford Nanopore Technologies (ONT) ONT sequencing은 분석하려는 double-stranded DNA에 어뎁터가 부착된 선형의 DNA molecule을 이용한다 [5]. 이들은 합성된 얇은 막 위에 있는 수백~수천 개의 나노 크기의 구멍에 들어가게 된다. 이 구멍에는 모터 단백질(motor protein)이 있는데 double-stranded DNA 끝에 결합하여 single-strand DNA를 만들어낸다. 이 single-strand DNA는 구멍을 통과하면서 전기신호를 발생시키는데 이를 측정하여 염기서열을 판독한다. 이 분석법은 1Mb 이상의 아주 긴 DNA 조각 (ultra-long read)들이 분석되기도 한다. 장비의 작은 크기로 인하여 휴대용이 가능하며 USB 포트를 전용 소프트웨어가 설치된 컴퓨터에 연결하면 분석이 가능하다는 장점이 있으나, 정확도가 일반적으로 87-98% 인 것으로 알려져 있다 [6].
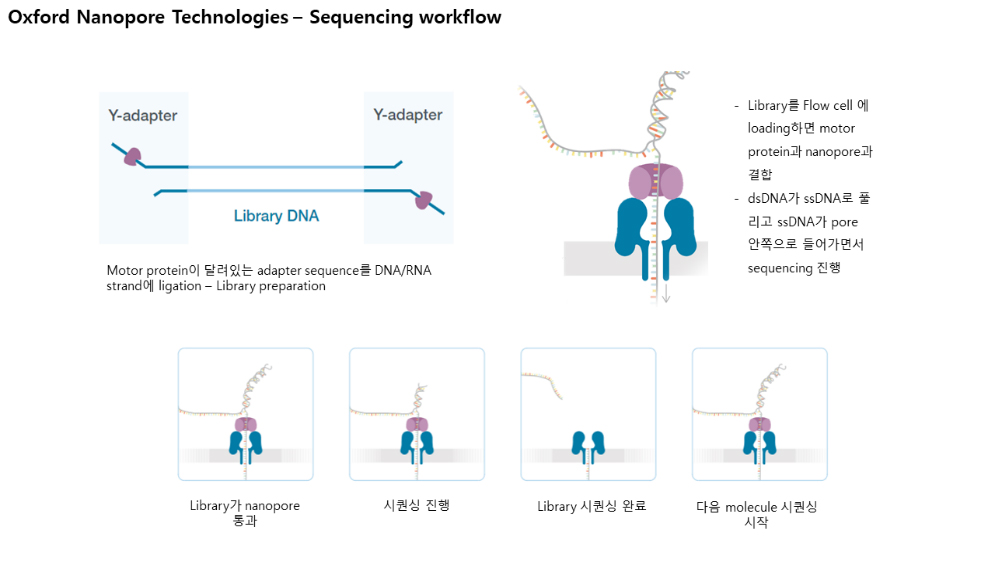 그림2. ONT 장비의 원리 (Oxford Nanopore Technologies사의 허가를 받아 사용
그림2. ONT 장비의 원리 (Oxford Nanopore Technologies사의 허가를 받아 사용
Long-read sequencing 검사의 활용 새로운 유전체 조립 (de novo assembly): 새로운 유전체 조립은 임의로 조각난 DNA 분자들의 염기 서열을 분석하고 이들의 끝과 끝을 맞추어 조각들을 차례대로 맞추면서 원래의 구조를 만들어가는 방법이다. 이들이 중간에 끊기지 않고 함께 연결되는 단위를 컨티그 (contig)라 한다. 일반적으로 한 개의 염색체는 한 개의 컨티그를 만드는 것이 이상적이다. 하지만 인간 유전체 안에는 반복염기 부위가 많기 때문에 sanger-based sequencing나 SR-NGS에 의해 유전체를 조립하게 된다면 수백만 개 이상의 컨티그가 발생한다. 반면 새로운 유전체 조립을 이용하면 긴 DNA 조각들을 정렬할 수 있어서 더 적은 수의 컨티그를 생성하므로 분석 시간이 단축된다. 특히 표준 염기서열이 없는 미생물 등에서는 새로운 유전체 조립이 필요한 경우가 많다 [7].
홑배수체형 위상 (haplotype phasing) 확인: 인간의 경우 두배수체형 (diploidy)의 유전체를 가지고 있기 때문에 변이나 염기서열의 위상을 확인하는 것이 중요할 수 있다. 예를 들어 상염색체 열성 질환과 연관된 유전자에서 유전자 기능에 영향을 주는 변이들은 같은 위상에 있는지 (cis-), 다른 위상에 있는지 (trans-) 확인해야 한다. 또한 HLA 유전자형은 이식 거부 반응 예측, 질병 연관성 연구 등에 중요한데, 이 유전자형을 결정하기 위해서는 위상을 확인할 수 있는 염기서열 분석이 중요하다. LRS는 같은 위상에서 한 번에 읽는 염기서열이 길기 때문에 유전자형을 더 정확하게 알 수 있다 [8].
구조적 변이 (structural variation) 확인: 구조적 변이는 50개 이상의 염기를 포함하는데, 기존의 SR-NGS 방법은 한 번에 분석 가능한 염기길이에 제한이 있으므로 이러한 변이는 검출하기 어렵다. 하지만 인간의 유전 질환에는 기존의 방법으로 원인이 밝혀지지 않은 구조적 변이들이 많다고 알려져 있다 [9]. 커다란 구조적 변이 (inversion, translocation, duplication, large indel)를 확인하기 위해서는 이 변이보다 더 큰 DNA 단일분자 조각을 한 번에 읽을 수 있어야 한다.
반복서열 확장 (tandem repeat expansion) 확인: 인간 유전체 안에는 많은 반복 염기서열 부위가 있고, 이 부위의 반복 수가 질환과 연관된 경우가 많다. 그런데 반복 수가 크게 증가하게 되면 짧은 길이의 염기서열 분석만으로는 이들의 반복 수를 측정하기 어렵다. 이럴 때 LRS가 도움이 될 수 있다. 예를 들어 뇌 신경 세포의 점진적 퇴화를 일으키는 헌팅턴 질환은 HTT 유전자의 ‘CAG’ 반복 수 증가로 인하여 나타나는데 LRS 방법을 통하여 측정이 가능하다 [10].
유사유전자 (pseudogene)의 구분: 유사유전자는 주로 유전자 복제 (gene duplication)나 역전위 (retrotransposition)로부터 발생한다. 이들은 코딩 유전자의 염기서열과 높은 상동성을 가지고 있으나 일부의 작은 염기서열 차로 인하여 기능을 가지지 못한다. 이러한 유사유전자는 많은 유전 질환의 분자유전 검사를 방해하는 요소로 작용한다. 예를 들어 선천성 부신과다 형성증 (congenital adrenal hyperplasia)과 관련있는 CYP21A2 유전자와 CYP21AP 유사유전자는 매우 유사한 상동성을 가지고 있다. 본 질환의 유전적 원인을 확인하기 위하여 CYP21A1P가 아닌 CYP21A2에 있는 변이를 확인해야 하는데 일반적인 SR-NGS 방법으로는 분석 되는 짧은 리드 (short read)가 두 유전자 중 어디에서 유래되었는지 알기 어렵다. 반면 LRS 분석은 유전자의 측부영역 (flanking region)을 포함하는 긴 리드 (long read)로 인하여 두 유전자 간의 구분이 수월할 수 있다 [11].
Long-read sequencing 검사의 전망 LRS 검사는 기존 SR-NGS 검사에 비해 높은 비용, 낮은 개별 염기서열의 정확도 및 낮은 처리율로 인하여 연구용으로 많이 사용되고 있다가 최근에 이러한 문제점이 점차 개선되면서 임상 진단에 활용이 늘고 있다. 또한 지금까지 DNA 연구 분석 위주로 기술했지만 RNA 연구 분석에도 도움이 될 수 있다 [12]. LRS를 이용하면 조직마다 다양하게 발현되는 full-length RNA isoform을 분석할 수 있고, 증폭 과정이 없기 때문에 생물학적으로 유의미한 발현 데이터를 만들 수 있다.
하지만 당장 SR-NGS 검사가 LRS 검사로 전환되기에는 어려운 점도 있다. 기존 LRS 검사의 문제점인 높은 비용, 낮은 정확도 및 낮은 처리율이 개선 되었다고 하지만 일반 검사로 하기에는 아직 부족하다. 그리고 기존의 생물정보학 (bioinformatics) 은 SR-NGS에 맞추어져 있고, LRS를 위한 분석에는 더 많은 개선이 필요하다. 또한 실제 LRS 검사를 진행해 본 경험상 실험 과정에서 SR-NGS 보다 까다로운 부분이 있었다. LRS 검사의 핵심은 긴 리드들을 분석하는 것인데 시퀀싱 장비에 들어가기 전 DNA를 준비하는 과정에서 이 긴 리드들이 절편화 (fragmentation)되지 않고 길게 유지하는데 특별한 주의가 필요했다. 또한 준비된 DNA를 시퀀싱 장비에 로딩하기 전 리드들의 길이 측정을 통한 질 관리 (quality control)가 중요한데 긴 리드들의 정확한 측정이 쉽지 않았다.
이러한 제한점들이 개선된다면 향후 유전체 분석은 하나의 참조 유전체 (reference genome)의 염기 서열과 다른 변이들을 확인하는 과정이 아니라 개개인의 유전체를 새로운 유전체 조립을 통하여 구성하고 완전한 일배체형의 염기서열을 분석할 수 있을 것이다.
[참고문헌]
1. Anderson, M.W. and I. Schrijver, Next generation DNA sequencing and the future of genomic medicine. Genes (Basel), 2010. 1(1): p. 38-69.
2. Mantere, T., S. Kersten, and A. Hoischen, Long-Read Sequencing Emerging in Medical Genetics. Front Genet,
2019. 10: p. 426.
3. Rhoads, A. and K.F. Au, PacBio Sequencing and Its Applications. Genomics Proteomics Bioinformatics, 2015. 13(5): p. 278-89.
4. Tang, L., Circular consensus sequencing with long reads. Nat Methods, 2019. 16(10): p. 958.
5. Bowden, R., et al., Sequencing of human genomes with nanopore technology. Nat Commun, 2019. 10(1): p. 1869.
6. Logsdon, G.A., M.R. Vollger, and E.E. Eichler, Long-read human genome sequencing and its applications. Nat Rev Genet, 2020. 21(10): p. 597-614.
7. Moss, E.L., D.G. Maghini, and A.S. Bhatt, Complete, closed bacterial genomes from microbiomes using nanopore sequencing. Nat Biotechnol, 2020. 38(6): p. 701-707.
8. Matern, B.M., et al., Long-Read Nanopore Sequencing Validated for Human Leukocyte Antigen Class I Typing in Routine Diagnostics. J Mol Diagn, 2020. 22(7): p. 912-919.
9. Merker, J.D., et al., Long-read genome sequencing identifies causal structural variation in a Mendelian disease. Genet Med, 2018. 20(1): p. 159-163.
10. Höijer, I., et al., Detailed analysis of HTT repeat elements in human blood using targeted amplification-free long-read sequencing. Hum Mutat, 2018. 39(9): p. 1262-1272.
11. Stephens, Z., et al., PB-Motif-A Method for Identifying Gene/Pseudogene Rearrangements With Long Reads: An Application to CYP21A2 Genotyping. Front Genet, 2021. 12: p. 716586.
12. Kovaka, S., et al., Transcriptome assembly from long-read RNA-seq alignments with StringTie2. Genome Biol, 2019. 20(1): p. 278.
