Nortable Research
Combining chromosome conformation capture and exome sequencing for simultaneous detection of structural and single-nucleotide variants
Technology Trend 학회뉴스씨젠의료재단
임상에서 유전성 질환이 의심될 때 임상에서 루틴으로 내는 검사들은 장단점이 명확하여 각 검사별로 검출하지 못하는 기전들이 있기 마련이다. 그렇다고 해서 whole genome long read sequencing등과 같은 발전된 기술들을 이용하자니 비용 문제가 걸린다. 따라서 보통 유전질환 환자를 진단하기 위해서는 의사가 환자의 표현형을 보고 질환의 원인이 되는 변이 종류를 예측하여 적절한 검사를 내야 한다. 그런데 이런 이상적인 진료가 이루어지려면 극도로 숙련된 전문가가 필요한 데다 그러한 전문가조차도 환자의 표현형만 보고서 항상 정확하게 유전자 병인을 집어낼 수 있는 것은 아니다. 따라서 현실적인 비용으로 다양한 종류의 유전자 변이를 한 번에 검출하는 검사가 필요하다. Hi-C assay를 비롯, chromosome conformation capture 법들은 proximity ligation techniques을 이용하여 유전체 전장에 걸친 염색질 접촉을 분석하는 방법이다. 저자들은 이전 연구에서 Hi-C map으로 역위, 전좌 등의 구조적 변이(SV)를 검출할 수 있음을 보여줌으로써 Hi-C의 적응증을 재정의하였다. 그러나 Hi-C로 SNV를 검출하려면 depth를 크게 늘려야 해서 다시 비용 장벽에 부딪히게 되었다. 따라서 본 논문에서 저자들은 임상에서 SV와 SNV를 효율적으로 검출할 수 있도록 chromatin conformation capture법에 기반한 새로운 방법을 개발하였다
저자들은 exome sequencing과 chromosome conformation capture를 통합한 접근법을 시도하여, Exo-C라고 이름붙였다. 간단히 설명하자면 우선 염색질을 고정 및 분리한다. 염색질을 DNase I나 S1 nuclease로 분해하고, proximity ligation 후 ligation junction과 exonic sequences를 가진 산물을 hybridization-based target capture panel로 enrich한다. 이렇게 구해진 시퀀스로 SV나 SNV 등등을 구하게 되며, 모든 Exo-C 데이터는 juicer toolbox 기반의 파이프라인을 통해 cool map과 hic 짝의 목록으로 정리한다.
각 검체에 대한 structural variants calling은 다음과 같이 시행한다. 본 글에서는 대표적이면서 가장 우수한 결과를 보인, 염색체 전좌를 검출하는 방법만 소개하기로 한다. 염색체 전좌를 calling하기 위해 gradient pattern search method와 line pattern sesarch method 두 가지 방법의 결과를 통합한다.
Gradient pattern search method는 Exo-C map에서 해상도별로 ‘dots of interest (DOIs)’를 Exo-C matrix의 원소로 설정한다. DOIs에 대한 검체와 콘트롤의 아래의 이항확률질량함수의 로그값의 차이가 문턱값 이하여야 한다.
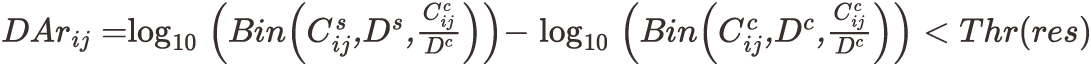
문턱값은 해상도별로 -4.0, -2.0, -1.0으로 다르게 설정한다. 각 DOI가 전좌의 절단점과 이웃한다라고 가정하여 절단점에서 거리가 1 bin, 2 bin,…이라고 가정한다. 그러면 DOI 근처의 모든 지점에 절단점으로부터의 거리에 따라 접촉확률 Part_cis(m)를 부여할 수 있다. 전좌가 없다고 가정하면, DOI 주변의 지점들은 Pcontrol_like 염색체간 접촉확률을 가질 것이다. DOI근처의 모든 지점에 대한 Pcontrol_like와 Part_cis(m)을 누적합산하면 DOI를 중심으로 한 사분면에 점들이 위치할 것이다. 각각의 사분면에 대해 1과 32 사이의 모든 k와 n 값에 대해 ArLG (Artifacts Level for Gradient pattern search) 확률을 계산한다.
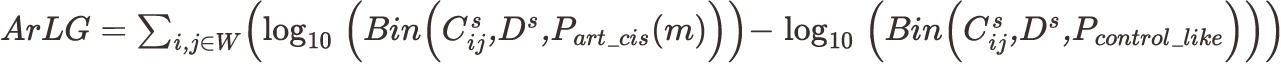
W는 window이고, Bin(Csij, Ds,Part_cis(m)는 좌표 (i,j)가 거리 m만큼 떨어진 지점과 cis-contact할 확률, Bin(Csij, Ds, Pcontrol_like는 좌표 (i,j)가 거리 m만큼 떨어진 지점과 trans-contact할 확률을 나타낸다. 각 DOI에서 가장 높은 ArLG 값이 나오는 window 하나만 기록하여 다음의 세 가지 filtering step을 적용한다: 1) ArLG>0 (cis-contact할 확률이 높음); 2) 어떤 DOI가 다른 DOI의 window에 포함될 때, ArLG가 높은 DOI만 남긴다; 3) log10(ArLG)>0.917*log10(contacts_sum)—0.92 (contact-sum은 윈도우 안에서 contact의 총합이다. 이 세 가지 필터를 모두 통과한 DOI와 그 윈도우는 전좌가 있는 것으로 간주한다.
Line pattern sesarch method는 DOI 설정까지는 동일하고, 각 DOI에 대해 다음과 같은 수식을 계산한다.
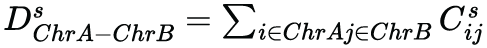
∑i∈ChrA 는 염색체 A에 속하는 Exo-C 열의 총합이다. Ds ChrA-ChrB와 Dc ChrA-ChrB는 각각 염색체 A와 B 짝에 속하는 검체와 컨트롤의 contact의 총합이다. 대조군 이항보다 표본 이항 모형에 속할 가능성이 낮은 열은 ArLL>0이다. 둘 사이의 거리가 5 bin 미만이고 ArLL>6일 때 두 열을 전좌로서 결합시킨다. 새로 생성한 열에서 ArLL을 재계산한다. 또 모든 열 조합에 대해서 동일한 계산을 수행하여 최댓값(삽입 지점에 가장 가까운 위치)를 찾는다. 마지막으로 검체 contact sum dependent 필터- log10(ArLL)>exp(1.73*log10(contacts_sum))-를 적용한다.
이 외에도 역위와 CNV 등 다양한 유전체 이상에 대한 수식이 있으나 기본적인 아이디어는 대동소이하다. 모든수식에 대한 결과들은 NGS나 SKY 등으로 구한 결과와 비교하여 평가하였다.
결과는 다음과 같이 나타내어진다. Exo-C로 검출한 염색체 전좌이다. 대각선 위에 위치하는 P136 검체에서 직교하는 프로파일을 관찰함으로써 염색체 4번과 9번 사이의 전좌를 확인할 수 있다. 컨트롤인 정상 염색체 P114에서는 해당 프로파일이 나타나지 않는다. Breakpoint의 위치 또한 좌표의 위치로써 확인할 수 있다.
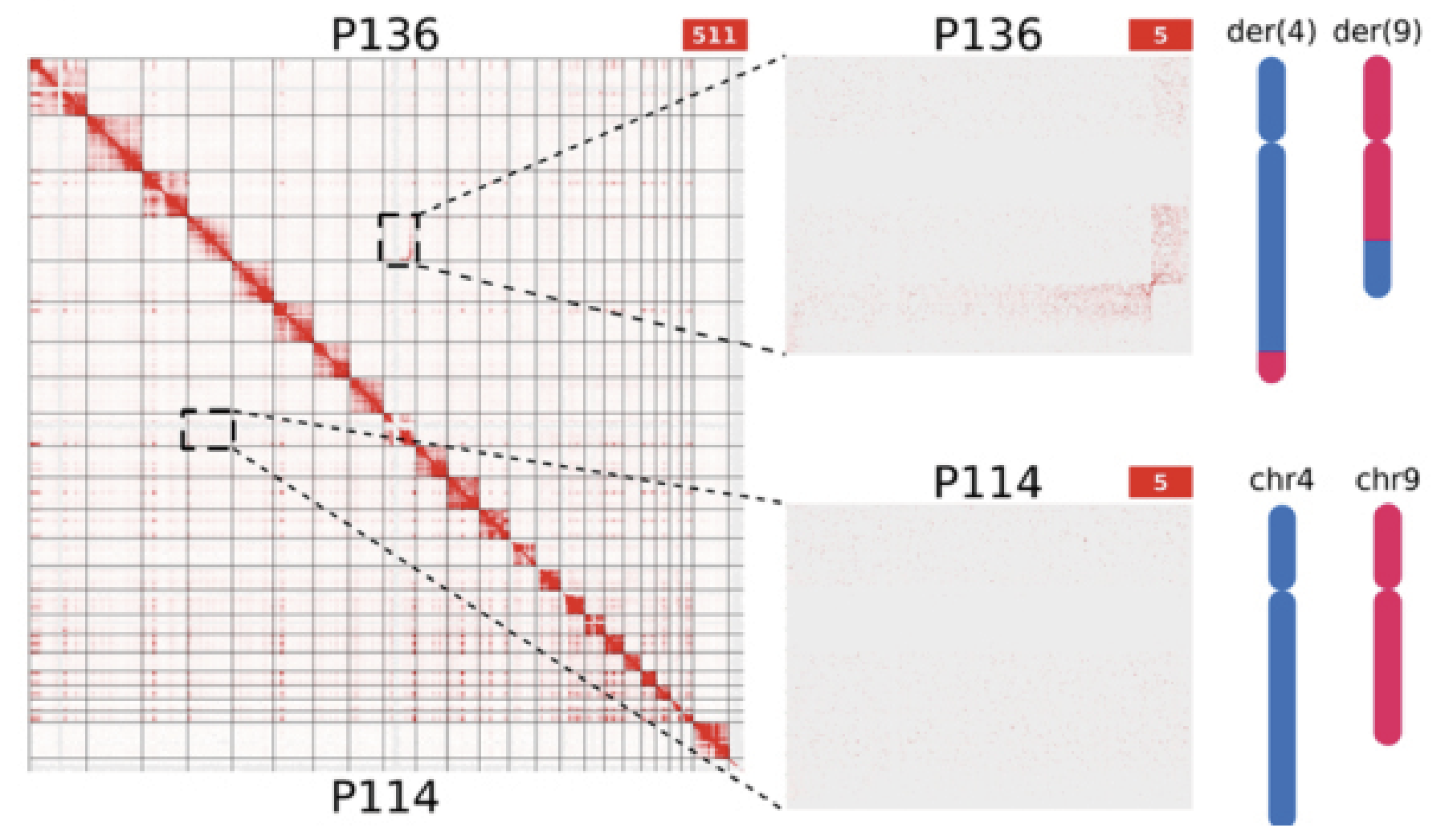
그림만 보면 시각적 변별력이 뛰어난 결과라고 생각되지만, Exo-C caller는 참값으로 생각되는 4가지 전좌를 모두 감지했을 뿐만 아니라 6가지 추가 콜을 생성했다. 따라서 위양성률이 높아서 40%의 낮은 정밀도를 나타냈다(재현율은 100%). 하지만 기존에 알려진 유전체 재배열 중 절반 이상이 500kb 미만의 작은 사이즈이기 때문에 본 연구의 낮은 정밀도는 과소평가된 값일 수 있다.

염색체 역위 검출에서는 역위가 0인 검체에서 21개의 위양성 역위 콜을 생성하였다. 정확도를 생각하면 형편없는 결과인 것 같지만 실제로 그 중 9 건은 기존에 알려진 결실이나 중복의 절단점과 일치했다. 이는 역위 콜러가 분류에서는 미흡한 점이 있지만 비정형 Hi-C 패턴을 가진 영역 자체는 꽤 정확하게 식별할 수 있다는 것을 시사한다. 따라서 Exo-C분석의 또 다른 유용성은 복잡한 염색체 재배열에서 절단점을 정확하게 짚어 냄으로써 pathogenicity를 입증할 수 있다는 점일 것이다. 왼쪽의 그림에서 검은 화살표는 절단점에서 Exo-C contact을 나타낸다.
본 연구에서 저자들은 chromosome conformation capture assay와 exome enrichment를 결합시킨 Exo-C가 표준 엑솜 시퀀싱과 유사한 비용으로 균형 전좌 및 역위 검출에 있어 기존 WES보다 우수함을 보여주었다. 기존의 방법을 개선하기 위해서 SV 호출에 중점을 두어 Exo-C 데이터 분석에 특화된 계산법을 개발한 것이 본 논문의 미덕이라고 할 수 있을 것이다. 이론상 기대되었던 바와 달리 inversion SV와 copy number variants(CNV), mosaic carrier의 분석은 추가적인 개선이 필요한 것으로 나타났다. 실제 임상과는 거리가 멀지만 비용으로 인한 검사 적용의 한계를 수학적인 방법으로 극복해보고자 하는 접근법이 신선하여 소개해보았다.
[References]
Gridina, M., Lagunov, T., Belokopytova, P. et al. Combining chromosome conformation capture and exome
sequencing for simultaneous detection of structural and single-nucleotide variants. Genome Med 17, 47 (2025).
https://doi.org/10.1186/s13073-025-01471-3
