고려대학교 바이오시스템의과학부
생명 현상을 이해하는 데 있어 세포라는 단위는 지극히 중요하다. 각 세포는 고유한 유전자 발현 양상을 지니고 있으며, 이러한 특성의 미세한 차이가 질병의 발생과 진행을 좌우한다. 연구자들은 오래전부터 유전체 정보를 활용해 질병의 원인과 병리기전을 규명하고자 했지만, 조직 전체의 평균적 발현 신호만을 얻는 전통적 분석기법으로는 세포 간 이질성을 세밀하게 구분하기 어려웠다. 바로 이 지점에서 단일세포 전사체 분석(single-cell transcriptome analysis)의 등장이 큰 전환점을 마련했다. 단일세포 분석 기술은 개별 세포 단위로 유전자 발현 프로파일을 얻어낼 수 있어, 동일한 조직 내에서도 각 세포 군이 지니는 고유 신호와 이를 둘러싼 상호작용 메커니즘을 파악할 수 있게 하였다. 이러한 혁신적 접근 방식은 암, 신경질환, 자가면역질환 등 다양한 의생명 분야에서 새로운 표적 발견 및 진단법 개발을 가속화하는 주요 동력이 되고 있다. 더욱이 단일세포 전사체 분석 기술이 최근 급격히 발전함에 따라, 한 번의 실험에서 수만에서 수십만 개 이상의 세포를 동시에 분석할 수 있는 대용량 데이터 생산이 가능해졌다.
본 기고문은 이러한 기술적 진보와 함께 급부상한 “싱글셀 아틀라스(single-cell atlas)”가 임상 유전체학과 맞춤형 치료 전략에 어떻게 접목될 수 있는지를 조망하려는 목적에서 작성되었다. 최근 단일세포 전사체 데이터를 대규모로 생산하고 공유하려는 움직임이 활발해지면서, 다양한 질환이나 조직에서 세포 유형을 총체적으로 분류하고, 각 세포 간 기능적 상호작용을 한눈에 파악할 수 있는 이른바 “세포 지도”가 속속 등장하고 있다. 이는 종양 생물학, 신경과학, 면역학 등 여러 분야에서 아직 미지의 영역으로 남아 있던 병리 기전을 분자 수준에서 추적 할 수 있게 만들었으며, 그 과정에서 발견되는 새로운 바이오마커나 치료 표적은 정밀의료 시대를 여는 핵심 자원이 되고 있다. 특히 특정 질환에서 핵심 역할을 하는 세포 유형이 무엇인지, 각 세포 안에서 어떤 유전자 네트워크가 작동하는지 등을 밝히는 연구가 잇따르면서, 임상의학은 환자 개인의 유전자적 배경과 미세한 조직·세포 변화를 종합적으로 고려하는 맞춤형 접근을 더욱 실현 가능하게 되었다. 본 기고문에서는 이러한 배경에서 싱글셀 아틀라스가 어떻게 구축되고 활용되는지, 그리고 임상 유전체학적 관점에서 어떤 새로운 가능성을 열어주고 있는지를 살펴보고자 한다.
단일세포 전사체 분석 기술이 보편화되면서, 여러 연구기관과 컨소시엄에서는 이 기술로부터 나온 방대한 데이터를 모으고, 이를 토대로 특정 조직 혹은 생물학적 상태에서의 세포 구성을 총체적으로 보여주는 “싱글셀 아틀라스”를 구축하고 있다. 단일세포 전사체 데이터는 세포 유형별 유전자 발현 양상을 정밀하게 포착하기 때문에, 과거 벌크 수준에서 놓쳤던 미세한 이질성과 기능적 차이를 체계적으로 보여줄 수 있다. 특히 질병과정에서 나타나는 특이적 세포 유형이나, 특정 변이가 실제로 작동하는 세포 집단을 찾는 데 매우 유용하기에, 연구자들은 일종의 형도를 만들 듯이 세포 지도를 그리고, 이를 임상적 해석에 활용한다. 그러나 이렇게 체계적 아틀라스를 구축하기 위해서는 비교 가능하고 호환성 높은 단일세포 데이터가 필수적이다. 각 연구마다 시료 준비, 시퀀싱 플랫폼, 분석 파이프라인이 다르기 때문에, 단순히 데이터를 모아놓는 것만으로는 공동 해석이 어려운 경우가 많다. 이 점에서 Human Cell Atlas(HCA)는 중요한 역할을 맡 고 있다 [1]. HCA는 이름만 보면 인간의 모든 세포 유형을 망라하는 아틀라스를 직접 제공하는 것처럼 보이지만, 실제로는 개별 연구자나 기관에서 생산된 데이터를 표준화된 형식으로 수집·공개하는 데 이터베이스의 성격을 지닌다. 즉, 전 세계 연구실에서 생성되는 단일세포 전사체 데이터를 동일한 메타정보 체계로 정리하고, 누구나 접근하여 재분석하거나 새로운 질환·조직 연구에 활용할 수 있도록 돕는다. 이는 대규모 국제 협업을 촉진하고, 연구자들이 더 쉽게 공동 연구를 펼칠 수 있는 기반을 제공한다. 이러한 표준화 덕분에, 단일세포 아틀라스를 구축하려는 연구자들은 HCA가 공개한 데이터를 가져와서, 특정 질환 맥락이나 특정 장기에 초점을 맞춘 새로운 아틀라스를 보다 효율적으로 만들 수 있게 되었다.
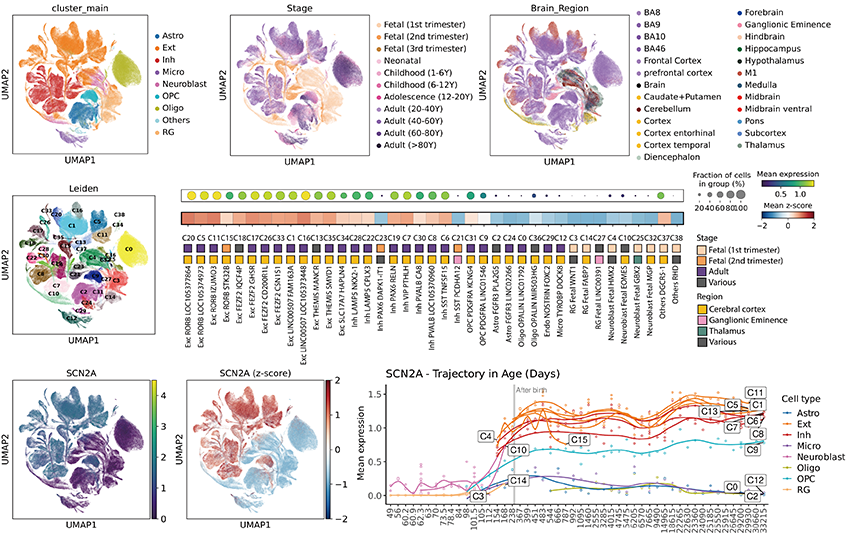 그림1
그림1
BTS 아틀라스 데이터 개요. 인간 뇌 발달 과정 및 세포 유형 별 SCN2A의 유전자 발현량
최근 다양한 아틀라스가 구축되며, 특정 질환이나 조직에 특화된 연구들이 진행되고 있다. Brain Transcriptome at the Single-cell Level (BTS) 아틀라스는 인간 뇌 발달 과정에서 특정 세포 유형과 유전자 발현 패턴을 분석하여 (그림 1), 신경질환과의 연관성을 연구하는 데 활용되고 있다[2]. Lung Cancer Cell Atlas는 폐암 환자의 종양 및 미세환경을 분석한 대규모 단일세포 전사체 데이터베이스로, 면역세포와 종양세포 간의 상호작용을 분석하고 폐암의 분자적 이질성을 탐색하는 데 사용되고 있다[3]. 또한, Single Cell Atlas of the Normal and Malformed Human Brain Vasculature는 정상 및 기형적 인간 뇌혈관 조직에서 세포 유형을 분류하고, 신경혈관 질환과 연관된 세포 변화를 추적하는 데 활용되고 있다[4].
유전체 연구가 발전하면서 질병에는 단일 유전자보다는 다수의 유전자가 관여한다는 사실이 밝혀졌다. 우리가 임상에서 보는 대다수의 질환들은 높은 수준의 유전적 이질성을 보인다. 동일한 임상적 진단을 받은 환자라도 각기 다른 유전자 변이를 가질수 있으며, 이에 따라 발병 연령, 증상의 양상, 치료 반응성이 달라질 수 있다. 그러나 개별적인 유전적 차이에도 불구하고, 특정한 공통된 표현형을 보이며 동일한 질환 범주로 분류된다. 이러한 현상은 단순한 우연이 아니라, 생물학적 시스템 내에서 공통적인 병리 기전이 존재하기 때문이다. 다양한 유전적 변이들이 센트럴 도그마 - DNA에서 RNA, 단백질 -로 번역되는 과정에서, 그리고 세포, 조직,기관의 순서로 발현되는 과정에서 종극에는 공통된 표현형을 형성한다. 이러한 개념을 기능적 수렴(functional convergence)이라고 한다.
신경발달장애의 일종인 자폐스팩트럼장애 (autismspectrum disorder; ASD)는 다양한 유전자가 연관되었다. 국제 공동연구를 통해 3만명 이상의 대규모 엑솜 유전체 데이터를 분석하였고, genome-wide association을 보이는 총 102개의 유전자를 규명하였다[5]. 하나의 질환에 관찰되는 다양한 유전자들은 기능적 수렴성이 존재할까? 초기의 사후 뇌조직 연구들은 이에 대한 답을 도출하였다. 뇌은행에 기증된 사후 뇌조직에서 벌크 전사체 데이터를 생산하여, 수정 후 8주 태아의 뇌에서부터 40세 성인의 뇌를 얻어, 총 16개의 뇌 부위에서 유전자 발현량 데이터를 생산했다. 이 데이터를 통해, 당시 ASD의 원인 유전자들을 활용하여, 유전자 네트워크를 조사하였고, 네트워크가 강하게 발현되는 발달 시점과 뇌 부위를 탐색했다. 그 결과, 임신중기 (수정 후 20주 내외)의 태아의 전전두엽피질(prefrontal cortex)에서 ASD 유전자의 네트워크가 집중적으로 나타남을 확인하였다[6]. 해당 발달시기는 인간 및 영장류의 뇌에서 고유하게 나타나는 피질의 확장이 일어나는 시점이며, 신경망을 복잡하게 형성한다. ASD 유전변이는 해당 시점과 부위에서 발현되는 유전자 네트워크에 영향을 미치고, 궁극적으로 뇌발달에 영향을 준다.
싱글셀 아틀라스는 세포 특이적인 발현량 정보를 확인하여, 보다 확장된 개념의 기능적 수렴성을 연구한다. BTS 아틀라스는 인간 사후 뇌조직에서 얻은 싱글셀 전사체 데이터를 활용하여, 총 39만개의 인간 뇌의 싱글셀 아틀라스를 구축하였다[2]. 이 데이터를 활용하여, ASD 원인 유전자들이 임신 중기의 전전두엽피질에서 신경세포가 발생하는 과정에서 높은 수준으로 발현하는 것을 확인하였다. 이러한 패턴은 신경세포에서만 특이적으로 관찰되며, 다른 신경질환들과는 다른 양상을 보인다. 예를 들어, 조현병(schizophrenia)의 경우, GWAS(Genome-wide association Study)를 통해 밝혀낸 common variant들은 방사형 아교세포(radialglia)에서 기능적 수렴성이 관찰된다. 그러나, 조현병의 rare variant는 흥분성 신경세포(excitatoryneuron)에서 주로 발현된다. 마찬가지로, 인간 뇌싱글셀 아틀라스를 활용하면 퇴행성 신경질환 원인 유전자들의 기능적 수렴을 규명할 수 있다[4]. 특정유전자 변이가 발현되는 세포 유형을 분석하면, 해당 변이가 신경세포의 기능에 미치는 영향을 이해할 수 있다. 또한, 싱글셀 아틀라스를 활용하면 퇴행성 신경질환에서 미세아교세포(microglia)나 희소돌기아교세포(oligodendrocytes)의 변화를 추적할 수 있으며, 특정 세포 유형에서 발병 기전이 어떻게 조절되는지를 밝히는 데 기여할 수 있다.
이러한 분석 결과는 특정 질병이 단순히 개별 유전자 변이로 설명되지 않고, 특정 세포 유형과 발달 시기에 따라 기능적으로 수렴하는 경향이 있음을 보여준다. 싱글셀 아틀라스를 활용하면 질환과 관련된 유전자 네트워크가 특정 발달 시점과 세포 유형에서 어떻게 작용하는지를 명확하게 규명할 수 있으며, 이는 맞춤형 치료법 개발에도 중요한 단서를 제공할 수 있다.
암은 단일한 질병이 아니다. 종양세포와 면역세포, 섬유아세포, 혈관세포 등이 복잡한 상호작용을 이루며 진행된다. 동일한 암 유형에서도 개별 환자의예후나 치료 반응이 크게 달라지는 이유는 종양의 미세환경 차이 때문이다. 특히, 암 미세면역환경은 면역세포의 구성과 활성 상태, 종양세포와의 상호작용 등에 따라 종양의 성장과 면역 회피 기전이 조절된다. 따라서, 종양 조직 내 개별 세포의 조성을 단일세포 수준에서 분석하는 것은 면역치료 반응성을 예측하고, 맞춤형 치료 전략을 수립하는 데 필수적이다. 싱글셀 아틀라스는 이러한 연구를 가능하게 하는 강력한 도구로, 종양 조직 내 다양한 세포 유형과 유전자 네트워크를 정밀하게 분석할 수 있도록 한다.
최근 대규모 싱글셀 아틀라스를 활용한 연구에서는 30가지 암 유형에서 490만 개 이상의 단일세포 전사체 데이터를 분석하여, 정상 조직과 종양 조직 간의 세포 유형 및 유전자 발현 패턴의 차이를 규명하였다[7]. 연구에 따르면, 종양 미세환경 내 특정면역세포 및 섬유아세포의 구성이 암의 진행과 면역치료 반응성을 결정하는 주요 요인으로 작용하며, 동일한 암 유형 내에서도 면역 미세환경이 크게 다를 수 있다는 사실이 밝혀졌다. 예를 들어, 폐암과 유방암에서 특정 섬유아세포 아형이 면역억제 환경을 조성하며, 이에 따라 면역치료 반응성이 달라진다. AKR1C1+ 염증성 섬유아세포는 CXCL1, CXCL3, CXCL8과 같은 염증성 사이토카인을 분비하여 면역억제 특성을 나타내는 반면, WNT5A+ 염증성 섬유아세포는 종양세포 및 기타 미세환경 요소와의 상호작용을 통해 암의 진행을 촉진하는 것으로 나타났다.
특정 면역세포 아형도 면역치료 반응성과 밀접한 연관이 있는 것으로 보고되었다. LAMP3+ 수지상 세포와 CCL19+ 섬유아세포는 면역치료 반응성이 높은 종양 환경에서 주로 발견되며, 이러한 세포들의 비율이 높을수록 면역관문억제제에 대한 반응성이 증가하는 것으로 나타났다. 또한, CD19+ B 세포 및 IGKC(high) 플라즈마 세포는 암 조직 내에서 면역 반응을 조절하는 주요 세포로 작용하며, 항체 매개 면역 반응을 통해 면역치료의 효과를 결정하는 요소로 작용할 가능성이 제시되었다. 이러한 연구 결과는 면역세포 및 섬유아세포의 특정 아형이 종양 미세환경 내에서 어떤 기능을 수행하는지를 이해하는 것이 암 치료 전략을 수립하는 데 필수적임을 시사한다.
GWAS는 질병과 연관된 유전 변이를 분석하는 강력한 방법으로 자리 잡았다. 그러나 GWAS에서 밝혀진 연관 변이 중 상당수는 단백질을 암호화하지 않는 논코딩 영역에 위치해 있어, 해당 변이의 기능적 의미를 해석하는 데 어려움이 있었다. Category-wide Association Study (CWAS)은 특정 세포 유형에서 특이적으로 발현되는 유전자의 조절 영역에 존재하는 변이들을 식별하는 방법이다[8]. 이 접근법은 싱글셀 아틀라스를 활용하여 특정 세포 유형에서 활발하게 발현되는 유전자의 프로모터나 인핸서 부위에 위치한 변이들이 특정 질병과 얼마나관련이 있는지를 평가한다. 예를 들어, CWAS를 이용한 연구에서는 ASD와 연관된 유전 변이들이 태아기 신경세포에서 활성화되는 조절 요소에 집중되어 있음을 발견했다. 또한, 알츠하이머병과 관련된 변이들이 특정 별아교세포(astrocytes)에서 발현되는 유전자의 조절 영역에서 확인되었으며, 이는 질병의 세포 특이적 기전을 이해하는 데 중요한 단서를 제공한다.
GWAS의 유전연관성을 싱글셀 데이터를 이용해 더욱 정밀하게 분석하는 방법도 개발되었다. 대표적인 예가 single-cell Disease Relevance Score(scDRS) 방법론이다[9]. scDRS는 특정 질병과 관련된 유전자들의 발현 패턴을 싱글셀 RNA 시퀀싱 데이터와 통합하여, 개별 세포 수준에서 질병과의 연관성을 평가하는 접근법이다. 먼저, GWAS 데이터를 기반으로 질병과 관련성이 높은 유전자 세트를 구축한 후, 해당 유전자들의 발현량을 바탕으로 개별 세포의 질병 점수를 산출한다. 이를 통해 특정 유전자 변이가 발현되는 주요 세포 유형을 식별하고, 해당 변이가 질병 발생과 어떻게 연결되는지를 분석할 수 있다.
이러한 접근법은 기존 GWAS 데이터만으로는 확인하기 어려웠던 세포 유형별 질병 연관성을 밝혀낼 수 있도록 돕는다. 특히, 논코딩 변이의 기능적 역할을 규명하는 데 싱글셀 아틀라스의 활용이 필수적이며, 기존 대량 시퀀싱 데이터로는 확인할 수 없었던 세포 특이적 조절 기전을 해석하는 데 기여할 수 있다. 앞으로 이러한 분석 방법이 더욱 발전하면서, 질병의 유전적 기전과 이를 조절하는 분자적 네트워크를 보다 정밀하게 규명할 수 있을 것으로 기대된다.
최근 인공지능 기술의 발전과 함께, 생명과학 분야에서도 대규모 데이터를 학습하여 다양한 생물학적 문제를 해결하는 파운데이션 모델이 등장하고 있다. 이러한 모델은 방대한 양의 데이터를 기반으로 사전 학습되며, 특정한 목적 없이 광범위한 패턴을 학습한 후 다양한 연구 문제에 적용될 수 있다는 점에서 기존의 데이터 분석 방식과 차별화 된다. 특히, 자연어 처리와 영상 인식 분야에서 성공적으로 활용된 딥러닝 기반 모델들이 싱글셀 전사체 데이터 분석에도 도입되면서, 생물학적 데이터 해석의 새로운 패러다임을 형성하고 있다.
대표적인 사례로 싱글셀 데이터 분석을 위한 대규모 언어 모델 기반 인공지능 시스템인 scGPT가 있다[10]. scGPT는 3,300만 개 이상의 단일세포 전사체 데이터를 학습하여, 세포 유형 간의 관계를 이해하고, 특정 세포 유형에서 발현되는 유전자 네트워크를 예측하는 기능을 갖춘다. 기존의 분석 방법들은 특정 실험에서 수집된 데이터를 바탕으로 제한적인 해석을 제공하는 반면, 이 모델은 다양한 연구에서 생산된 데이터를 통합하여 일반화된 생물학적 규칙을 도출할 수 있다. 이를 통해 새로운 세포 유형을 자동으로 식별하거나, 특정 질병과 연관된 유전자 세트를 입력하면 해당 유전자가 가장 활발하게 발현되는 세포 유형을 예측할 수 있어 질병 연구와 신약 개발에 중요한 단서를 제공한다.
생명과학 연구는 이제 개별 세포 수준에서 생물학적 과정을 해석하는 단계로 전환되고 있다. 싱글셀 아틀라스는 기존 유전체학이 제공하지 못했던 세포 유형별 유전자 발현 패턴을 정밀하게 분석할 수 있도록 하며, 이를 통해 질병의 병리적 기전을 보다 세밀하게 규명할 수 있다. 그러나 이러한 발전에도 불구하고, 여전히 해결해야 할 기술적 과제들이 남아있다.
첫째, 싱글셀 데이터를 연구마다 다른 실험 조건과 분석 방법으로 생성하는 현재의 방식은 연구 간 비교 가능성을 저해한다. 데이터의 표준화와 분석 파이프라인의 통합이 필수적이며, 이를 통해 보다 신뢰할 수 있는 결과를 도출해야 한다. 둘째, 단일세포 분석이 조직 내 세포 간 상호작용과 공간적 정보를 충분히 반영하지 못하는 한계를 극복하기 위해, 공간 전사체 분석과의 융합이 필요하다. 셋째, 대규모 임상 데이터를 싱글셀 분석과 연계하여, 연구 결과를 실제 환자 맞춤형 치료로 적용할 수 있는 정밀 의료 모델 구축이 요구된다.
향후 싱글셀 아틀라스는 더 많은 데이터를 포함하고 학습하여, 개인 간 유전적 차이를 반영하는 수준으로 발전할 것이다. 현재의 아틀라스는 조직과 질병 유형별로 데이터를 통합하는 단계에 머물러 있지만, 궁극적으로는 환자 개개인의 세포 특성을 예측하고, 질병의 발병 가능성을 사전에 평가하는 도구로 활용될 가능성이 크다. 이를 위해, 더욱 다양한 인구집단과 환경 요인을 포함하는 데이터 확장이 필요하며, 인공지능 및 기계학습을 활용한 해석 기법의 발전도 필수적이다.
싱글셀 아틀라스는 단순한 데이터베이스가 아니다.그것은 생물학적 복잡성을 해석하는 새로운 방식이며, 우리가 질병을 진단하고 치료하는 방식을 근본적으로 변화시킬 도구이다. 이 연구가 어디까지 확장될지는, 우리가 그것을 어떻게 발전시키는가에 달려 있다.
[References]
1. Regev, A., et al., The Human Cell Atlas. Elife, 2017. 6.
2. Kim, S., et al., An integrative single-cell atlas for exploring the cellular and temporal specificity of genes related to
neurological disorders during human brain development. Exp Mol Med, 2024. 56(10): p. 2271-2282.
3. Salcher, S., et al., High-resolution single-cell atlas reveals diversity and plasticity of tissue-resident neutrophils in
non-small cell lung cancer. Cancer Cell, 2022. 40(12): p. 1503-1520 e8.
4. Winkler, E.A., et al., A single-cell atlas of the normal and malformed human brain vasculature. Science, 2022.
375(6584): p. eabi7377.
5. Satterstrom, F.K., et al., Large-Scale Exome Sequencing Study Implicates Both Developmental and Functional
Changes in the Neurobiology of Autism. Cell, 2020. 180(3): p. 568-584 e23.
6. Willsey, A.J., et al., Coexpression networks implicate human midfetal deep cortical projection neurons in the
pathogenesis of autism. Cell, 2013. 155(5): p. 997-1007.
7. Kang, J., et al., Systematic dissection of tumor-normal single-cell ecosystems across a thousand tumors of 30
cancer types. Nat Commun, 2024. 15(1): p. 4067.
8. Kim, Y., et al., CWAS-Plus: estimating category-wide association of rare noncoding variation from whole-genome
sequencing data with cell-type-specific functional data. Brief Bioinform, 2024. 25(4).
9. Zhang, M.J., et al., Polygenic enrichment distinguishes disease associations of individual cells in single-cell RNAseq
data. Nat Genet, 2022. 54(10): p. 1572-1580.
10. Cui, H., et al., scGPT: toward building a foundation model for single-cell multi-omics using generative AI. Nat
Methods, 2024. 21(8): p. 1470-1480.
